Article
Generics - equal or not?
- Donald J. Birkett
- Aust Prescr 2003;26:85-7
- 1 August 2003
- DOI: 10.18773/austprescr.2003.063
Generic products must be bioequivalent to the innovator brand before they can be marketed in Australia. There are no generic formulations of drugs with a narrow therapeutic index as it would be difficult for them to meet the required standard of bioequivalence. In Australia most generic drugs are marketed with a brand name. Some generic brands are manufactured by the same company that produces the innovator brand of the drug. Although generic brands are usually cheaper the proliferation of brands may cause confusion.
From time to time, controversies and claims arise regarding generic prescribing and generic substitution.
For example, a support group for people with epilepsy issued a news release that stated:
These concerns make it worthwhile to revisit the issues and to try and sort fact from opinion and fiction.
The term 'generic product' is used in different ways. It can mean a product marketed under the drug's non-proprietary approved name, or it can, as is usual in Australia, mean a product marketed under a different brand (proprietary) name. It is sometimes used to mean any product from a company other than the innovator (research-based) manufacturer.
A common use of the term (and that used by the World Health Organization (WHO)), is for a pharmaceutical product that is:
In Australia writing the non-proprietary (generic) name on a prescription allows the pharmacist to dispense any brand of the drug. The pharmacist does not have to dispense the cheapest brand.
This policy enables the pharmacist, without reference back to the prescriber, to dispense a different brand of the drug even though the doctor has written a prescription for a particular brand. In Australia, doctors can endorse the prescription to prevent substitution.
Two pharmaceutical products are bioequivalent if they are pharmaceutically equivalent and their bioavailabilities (rate and extent of availability) after administration in the same molar dose are similar to such a degree that their effects, with respect to both efficacy and safety, can be expected to be essentially the same. Pharmaceutical equivalence implies the same amount of the same active substance(s), in the same dosage form, for the same route of administration and meeting the same or comparable standards.
Product quality and bioequivalence data are required before a generic product can be registered in Australia or listed on the Pharmaceutical Benefits Scheme (PBS). The quality data required include purity, stability, good manufacturing practice and quality control. These data are the same as those required for innovator products. It has sometimes been suggested that generic products may contain ratios of enantiomers (optical isomers) that are different from the innovator product. This argument cannot be sustained, as conventional chemical synthesis of the active drug produces a racemic (equal) mixture of the two enantiomers. Data on the enantiomeric ratio of the active substance in a generic product would in any case be required before registration in Australia.
Bioequivalence is usually assessed by single dose in vivo studies in healthy volunteers. The reference product is usually the innovator product that is marketed in Australia, but for older drugs it may be another generic that is the market leader in Australia. Figure 1 shows a simulation of such a study.
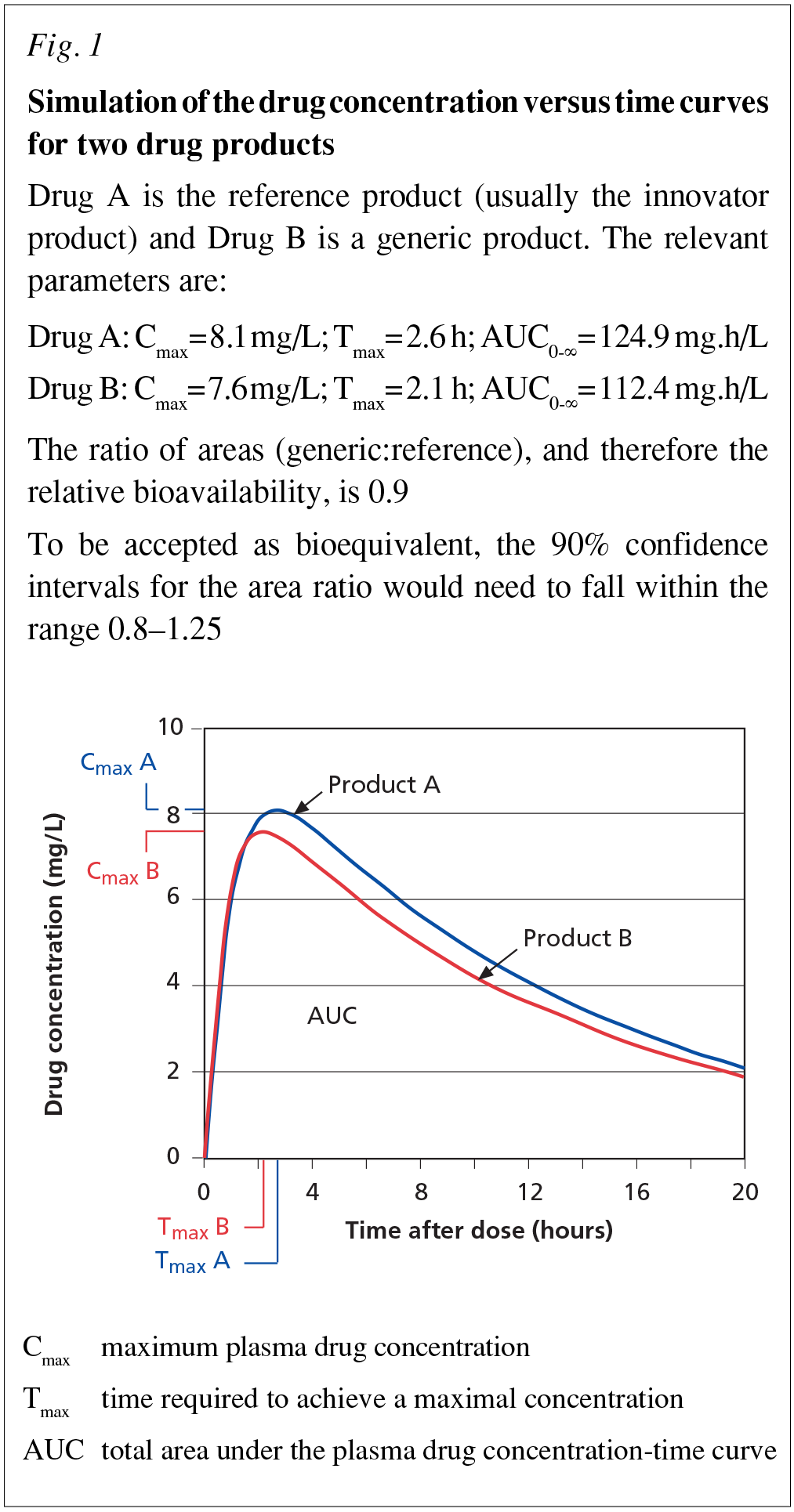
The regulatory limits applied are that the 90% confidence intervals for the ratios (test:reference) of the areas under the drug concentration versus time curves (AUC ratio) and the maximum plasma drug concentrations (Cmax ratio) must fall between 80% and 125%. (The confidence limits are asymmetrical because log transformed data are used in the comparison.) The times to maximum plasma concentration (Tmax) for the test and reference product should also be similar. These requirements for similarity between the two products are therefore in both the extent of absorption (AUC ratio) and the rate of absorption (Cmax and Tmax ratios). In addition, most regulatory authorities would look at the intersubject variability for the two products and ask questions if there was a marked difference between them. Products satisfying the bioequivalence requirements can reliably be assumed to produce similar clinical effects when used interchangeably in the same patient.
It is sometimes claimed that the 80 to 125% limit means there can be a 45% variation between the new product and the reference product, but this is not really the case. The average ratio (point estimate) is usually reasonably close to 100% and this is the value of maximum likelihood for the comparison. If the average ratio is close to the 80 or 125% regulatory limits then the data would have to be very tight indeed to prevent the 90% confidence intervals falling outside the regulatory boundaries.
Commonly there are a number of generic products linked by a 'chain of inference'. For example, two brands x and y may both have been shown to be bioequivalent to the market leader brand z. Can brands x and y then be considered bioequivalent? They have not been directly compared in a formal bioequivalence study, but in practical terms would be very unlikely to fail if directly compared. The pragmatic decision is taken to consider all brands interchangeable. It would be practically and financially very difficult, and ethically unacceptable, to require each brand to be compared with every other brand in formal human studies.
For a drug with a narrow therapeutic index and/or with saturable metabolism it may well be appropriate to require tighter bioequivalence limits for generic products. In fact, there are no generic products in Australia, for example, for digoxin (narrow therapeutic index and established bioavailability problems) or for phenytoin (saturable metabolism, narrow therapeutic index and bioavailability problems). The problems in the early 1970s that focused attention on phenytoin bioavailability occurred when there was a change of excipient (from calcium sulphate to lactose) in the innovator formulation. Another drug with a narrow therapeutic index is warfarin. There are two warfarin brands on the market in Australia, but there has been no formal bioequivalence comparison made of them so they are not interchangeable.
Establishing bioequivalence for interchangeable controlled-release products usually requires more extensive data including clinical trial data. However, some of these products are available in Australia (e.g. enteric-coated sodium valproate, sustained-release verapamil and controlled-release diltiazem).
Bioequivalence issues are not confined to generic products. The clinical trial data on which marketing of the innovator product is based are usually obtained with formulations which differ from that ultimately marketed. The requirements for establishing bioequivalence between trial and marketed formulations are similar to those needed when assessing generics. Furthermore, innovator (and generics) manufacturers will frequently change their manufacturing process or site of manufacture and are required to show by appropriate in vitro or in vivo studies that bioavailability has not changed. The data which link an innovator market formulation back to the clinical trial data are therefore essentially the same as those required to establish interchangeability for generic products.
One valid concern in relation to generics is that individual patients could have idiosyncratic sensitivity to excipients such as colourings that are in the generic but not innovator product. This can occur, but is very rare and is not a problem limited to generics. Changes of excipients in innovator products could cause similar adverse effects.
The Therapeutic Goods Administration evaluates all products that are intended to be interchangeable. It assesses them for quality and bioequivalence with the Australian innovator or market leader product. This usually requires in vivo bioequivalence data but, if satisfactorily justified by the sponsor, may be based on in vitro dissolution data for drugs with no known bioavailability problems.
Interchangeable products are marked in the Schedule of Pharmaceutical Benefits by a letter (a or b) and brand substitution by the pharmacist is permitted, unless the prescriber has indicated otherwise on the prescription. A brand premium, paid by the patient, is charged if the pharmacist dispenses a brand which costs more than the base-priced brand.
There has recently been a large proliferation in the number of 'generic' brands available through the PBS. This has resulted from the marketing of brands named according to the pharmacy chain selling them (e.g. Chem mart, Gen Rx, health sense, Terry White Chemists). They are, in fact, all exactly the same product made by the same manufacturer and just packed and branded (named) differently. This unnecessary proliferation of brands is unfortunate and has the potential to cause confusion, but cannot be prevented under current legislation. A similar twist applies to some of the oral contraceptive products. Some manufacturers have marketed the innovator product under a different brand name as an interchangeable 'generic'. This allows a premium (of the order of $7-9) to be charged for the original 'innovator' product which is then strongly promoted.
There is no evidence in Australia that generic drugs are dangerous and impair the safety and efficacy of treatment. Our regulatory regime is world standard and conforms to requirements in regions such as Europe and the USA. Indeed, the Australian generic and bioequivalence requirements are 'harmonised' to those in Europe. There is also no evidence of systematic problems occurring because of generic availability and substitution. On the other hand, generics are cost-saving and allow the drug and health budgets to be spread further to enable access to new and expensive treatments where these offer cost-effective health outcomes.
Birkett DJ. Pharmacokinetics made easy. 2nd ed. Sydney: McGraw-Hill Australia; 2002.
Professor Birkett is now Executive Director, Research and Development at Johnson & Johnson Research Pty Ltd.
The following statements are either true or false.
1. In Australia, generic drugs must be bioequivalent to the innovator or market-leading brand of the drug.
2. The two brands of warfarin in Australia are not interchangeable.
Answers to self-help questions
1. True
2. True
Professor, Department of Clinical Pharmacology, Flinders University and Flinders Medical Centre, Adelaide
