SUMMARY CMI
AROPAX®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I using AROPAX?
AROPAX contains the active ingredient paroxetine (as hydrochloride hemihydrate).
AROPAX is used to treat depression, panic attack prevention, anxiety and other illnesses by helping to control mood.
For more information, see Section 1. Why am I using AROPAX? in the full CMI.
2. What should I know before I use AROPAX?
Do not use if you have ever had an allergic reaction to AROPAX or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use AROPAX? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with AROPAX and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use AROPAX?
- The usual dose of AROPAX for depression, social anxiety disorder/social phobia, generalised anxiety disorder or post-traumatic stress disorder is one 20 mg tablet per day.
- To treat obsessions and compulsions or panic attacks, the usual dose of AROPAX is two 20 mg tablets per day.
- AROPAX tablets can be broken in half but should not be chewed.
- AROPAX should be taken in the morning, preferably with food.
More instructions can be found in Section 4. How do I use AROPAX? in the full CMI.
5. What should I know while using AROPAX?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Drinking alcohol |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using AROPAX? in the full CMI.
6. Are there any side effects?
Like other medicines, AROPAX can cause some side effects. If they occur, they are most likely to be minor and temporary.
Mild side effects can include feeling sick, dry mouth, constipation, decreased appetite, diarrhea, vomiting, drowsiness, dizziness, difficulty in getting to sleep, impaired sexual function, weakness, feeling sweaty or shaky, bruising, abnormal dreams (including nightmares), weight gain.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI
FULL CMI
AROPAX®
Active ingredient(s): paroxetine (as hydrochloride hemihydrate)
Consumer Medicine Information (CMI)
This leaflet provides important information about using AROPAX. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using AROPAX.
Where to find information in this leaflet:
1. Why am I using AROPAX?
2. What should I know before I use AROPAX?
3. What if I am taking other medicines?
4. How do I use AROPAX?
5. What should I know while using AROPAX?
6. Are there any side effects?
7. Product details
1. Why am I using AROPAX?
AROPAX contains the active ingredient.
AROPAX belongs to a group of medicines called selective serotonin reuptake inhibitor (SSRI) antidepressants. They are thought to work by their action on brain chemicals called amines which are involved in controlling mood.
AROPAX is used to treat:
- Depression which is longer lasting or more severe than the ‘low moods’ that everyone has from time to time. It is thought to be caused by a chemical imbalance in part of the brain that affects your whole body and can cause emotional and physical symptoms. You may feel low in spirit, lose interest in usual activities, have poor appetite, disturbed sleep, wake up early, have low energy and feel guilty over nothing.
- Irrational fears or obsessional behaviour: These can also be due to chemical imbalance in parts of the brain.
- Panic attacks: AROPAX may be used to help prevent panic attacks.
- Patients who may avoid and/or are fearful of social situations.
- Patients with excessive anxiety and worry, and who feel irritable, restless and/or tense in the muscles.
- Repetitive and distressing recollections of a past traumatic event.
2. What should I know before I use AROPAX?
Warnings
In some children and adolescents, treatment with an antidepressant increases suicidal thinking or actions.
- It is important to discuss all the risks of treating depression and also the risks of not treating it. You should discuss all treatment choices with your doctor, not just the use of antidepressants.
- Patients (and caregivers of patients) need to monitor for any worsening of their condition and/or the emergence of thoughts of suicide or suicidal behaviour or thoughts of harming themselves and to seek medical advice immediately if these symptoms present (see Section 2. What should I know before I use AROPAX - Children and Adolescents).
There is an increased risk of breaking a bone in people taking medicines like AROPAX. This risk is greatest during the early stages of treatment.
Do not use AROPAX if:
- you are allergic to paroxetine, or any of the ingredients listed at the end of this leaflet. Always check the ingredients to make sure you can use this medicine.
- you are pregnant or intend to become pregnant.
- you have taken AROPAX before and became unwell, tell your doctor or pharmacist before taking the first dose.
- you are taking any other medications for the treatment of depression or have done so in the last 2 weeks. Taking AROPAX with another antidepressant may cause a serious reaction.
- You must not take AROPAX until 2 weeks after stopping monoamine oxidase inhibitor drugs (MAOIs). Examples of MAOIs are phenelzine and tranylcypromine. Another MAOI includes the antibiotic linezolid. There may be others so please check with your doctor. Taking AROPAX with a MAOI may cause a serious reaction.
- you are taking or have recently taken (within the last two weeks) a medicine called methylthioninium chloride (methylene blue).
- you are taking thioridazine for the treatment of schizophrenia.
- you are taking pimozide.
- the expiry date (EXP) printed on the pack has passed.
- the packaging is torn or shows signs of tampering.
Check with your doctor if you:
- you are allergic to foods, dyes, preservatives or any other medicines.
- have any other medical conditions such as:
- liver problems
- heart problems
- kidney problems
- epilepsy
- mania
- raised pressure in the eye
- problems with blood clotting
- other psychiatric conditions (bipolar disorder)
- diabetes
- history of bleeding disorders, such as after childbirth - you are pregnant or intend to become pregnant.
- you are breastfeeding or wish to breastfeed.
Your doctor will discuss with you the possible risks and benefits of using AROPAX during breastfeeding.
The signs of an allergic reaction are wheezing, swelling of the lips/mouth, difficulty in breathing, hayfever, lumpy rash (hives) or fainting.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
- If you take AROPAX near the end of your pregnancy, there may be an increased risk of heavy vaginal bleeding shortly after birth, especially if you have a history of bleeding disorders.
- Your doctor or midwife should be aware that you are taking AROPAX so they can advise you.
- Studies show that use of paroxetine in early pregnancy (first 13 weeks) may be associated with an increased risk of some birth defects in babies.
- If you become pregnant or intend to become pregnant while taking paroxetine, you should make an appointment to see your doctor and have your treatment reviewed.
- It is important that you do not stop taking paroxetine suddenly.
- Paroxetine is a medicine that can have withdrawal side effects if stopped suddenly.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
Fertility
Medicines like AROPAX may affect your sperm. Fertility in some men may be reduced while taking AROPAX.
Elderly
Take special care with AROPAX if you are over 65 years of age as AROPAX may cause a reduction in the amount of sodium within your blood which can lead to sleepiness and muscle weakness. If you experience these symptoms, please consult your doctor as soon as possible.
Children and adolescents
AROPAX is not recommended for use in children and adolescents under 18 years.
- The use of AROPAX is not recommended to treat depression in children and adolescents under 18, as the drug has not been shown to be effective in this age group and there are possible unwanted effects.
- Information from clinical trials has suggested that young adults, particularly those with depression, may be at an increased risk of suicidal behaviour (including suicide attempts) when treated with AROPAX, especially during initial treatment (generally the first one to two months).
- The majority of attempted suicides in clinical trials in depression involved patients aged 18 to 30 years.
- Family and caregivers of children and adolescents being treated with antidepressants for major depressive disorder or for any other condition (psychiatric or non-psychiatric) need to monitor them for the emergence of agitation, irritability, unusual changes in behaviour, as well as the emergence of thoughts of suicide, and to report such symptoms immediately to their doctor.
- It is particularly important that monitoring be undertaken during the initial few months of antidepressant treatment or at times of dose increase or decrease.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with AROPAX and affect how it works. In particular tell your doctor if you are taking any of the following medicines which:
- treat depression, anxiety, schizophrenia or attention-deficit hyperactivity disorder (ADHD) such as tryptophan, hypericum perforatum (St John's Wort), perphenazine, risperidone, lithium or atomoxetine.
- are used in anaesthesia or to treat pain or chronic pain, specifically tramadol or fentanyl.
- lower blood pressure or treat heart conditions, such as metoprolol or flecainide.
- control epilepsy (anti-convulsants), such as phenytoin, carbamazepine, phenobarbital.
- thin blood (anti-coagulants), such as warfarin, aspirin, non-steroidal anti-inflammatory drugs (NSAIDs).
- treat Parkinson's disease, such as selegiline, procyclidine.
- treat stomach ulcers, such as cimetidine.
- treat migraine attacks such as sumatriptan.
- treat or prevent breast cancer, specifically tamoxifen.
- treat HIV infection such as a combination of fosamprenavir and ritonavir.
- Used in anaesthesia, such as mivacurium and suxamethonium.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect AROPAX.
4. How do I use AROPAX?
How much to use
- The usual dose of AROPAX for depression, social anxiety disorder/social phobia, generalised anxiety disorder or post-traumatic stress disorder is one 20 mg tablet per day. Your doctor may increase the dose slowly over several weeks. This may require you to break the tablet in half.
- To treat obsessions and compulsions or panic attacks, the usual dose of AROPAX is two 20 mg tablets per day. Your doctor may start you on a lower dose (half a tablet) and increase the dose slowly over several weeks. This may require you to break the tablet in half.
- Follow the instructions provided and use AROPAX until your doctor tells you to stop.
When to take AROPAX
- AROPAX should be taken in the morning, preferably with food.
How to take AROPAX
- Take AROPAX with a full glass of water or another liquid.
- AROPAX tablets can be broken in half but should not be chewed.
Instructions for use:
- An extra protective packaging layer is used for this medicine. Tablets will need to be firmly pushed out through the foil to remove them from the blister.
How long to take AROPAX
Keep taking your AROPAX for as long as your doctor tells you.
Your doctor may decide that you should continue to use AROPAX for some time, even when you have overcome your problem. For best effect AROPAX must be taken regularly.
Your doctor will tell you when and how AROPAX should be discontinued.
Your doctor will usually recommend that you stop treatment by slowly reducing the dosage over a period of several weeks. When you stop treatment with AROPAX, especially if this is done suddenly, you may experience unwanted symptoms. Please see the Section 6. Are there any side effects?
If you forget to use AROPAX
- Do not take an extra dose. Wait until the next day and take your normal dose then.
- Do not try to make up for the dose that you missed by taking more than one dose at a time.
- If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
- Do not take a double dose to make up for the dose you missed.
If you use too much AROPAX
If you think that you have used too much AROPAX, you may need urgent medical attention.
If you take too much AROPAX, symptoms include sedation, blood pressure changes, involuntary muscle contractions and facial flush. It rarely can lead to seizures, heart rhythm changes and unconsciousness. For more information, see Section 6. Are there any side effects?
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using AROPAX?
Things you should do
Call your doctor straight away if you:
For any reason, have not taken your medicine exactly as directed. Otherwise, your doctor may think that it was not working as it should and change your treatment unnecessarily.
Like other drugs of this type, AROPAX will not relieve your symptoms straight away. People generally start feeling better in a few weeks or so. Occasionally, the symptoms of depression or other psychiatric conditions may include thoughts of harming yourself or committing suicide. It is possible that these symptoms may continue or increase until the full antidepressant effect of your medicine becomes apparent.
All thoughts of suicide must be taken seriously. Tell your doctor immediately or go to the nearest hospital if you have any distressing thoughts or experiences during this initial period or at any other time.
Also contact your doctor if you experience any worsening of your depression/other symptoms at any time during your treatment.
Tell your doctor or go to the hospital immediately if you experience symptoms of serotonin syndrome or neuroleptic malignant syndrome, which are rare life-threatening conditions. These symptoms include high body temperature, stiffness, involuntary muscle jerks, confusion, extreme agitation, delirium and coma.
Remind any doctor, dentist or pharmacist you visit that you are using AROPAX.
Things you should not do
- Do not stop using this medicine suddenly.
- Do not give this medicine to anyone else, even if their symptoms seem similar to yours.
- Do not use AROPAX to treat any other complaints unless your doctor says to.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how AROPAX affects you.
Tests have shown that AROPAX does not have a marked effect on driving ability. However, AROPAX may cause drowsiness, dizziness or lightheadedness in some people. Make sure you know how you react to AROPAX before you drive a car or operate machinery.
Drinking alcohol
Tell your doctor if you drink alcohol.
Although drinking moderate amounts of alcohol is unlikely to affect your response to AROPAX, it is best to avoid alcohol while you are taking this medicine.
Looking after your medicine
- Keep your tablets in the blister pack until it is time to take them.
- Keep the pack in a cool dry place.
- Do not leave it in the car on a hot day.
- Do not store medicine in the bathroom or near a sink.
Heat and dampness can destroy some medicines.
Keep it where young children cannot reach it, such as in a locked cupboard.
Stopping treatment
Do not stop taking AROPAX even if you begin to feel better.
Your doctor may decide that you should continue to use AROPAX for some time, even when you have overcome your problem. For best effect AROPAX must be taken regularly.
Your doctor will tell you when and how AROPAX should be discontinued.
When your doctor decides that you should stop taking AROPAX the dose may be reduced slowly or the time between doses increased over 1 or 2 weeks. Some people may have symptoms such as dizziness, anxiety, sleep disturbances, pins and needles, electric shock sensations or feeling sick and sweating if AROPAX is stopped, particularly if stopped suddenly.
Although AROPAX is not recommended for children under 18 years of age, additional symptoms that have been experienced by children whilst stopping treatment are abdominal pain, nervousness and mood changes.
Please see additional information under Section 6. Are there any side effects?
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Other rare events that have been reported with AROPAX include:
- blurred vision
- abnormal liver function
- low levels of sodium in the blood, especially in older people
- bleeding disorders, including nose bleeds and gastrointestinal bleeding which occurs very rarely
- hormone disturbances
- mood of excitement, over-activity and uninhibited behaviour
- confusion
- seizures
- rash caused by light
- itchy rash, hives, swelling of the face, lips, mouth, tongue or throat
- akathisia (restlessness or difficulty keeping still, caused by medicines to treat mental disorders).
- irresistible urge to move the legs (Restless Legs Syndrome)
- menstrual period disorder (including heavy periods, bleeding between periods and absence of periods.
- heavy vaginal bleeding shortly after birth.
- severe allergic reactions.
Unwanted effects that may occur on stopping treatment:
Symptoms may include
- dizziness
- sensory disturbances such as, pins and needles, burning sensations and electric shock-like sensations
- sleep disturbances, including intense dreams
- agitation or anxiety
- feeling sick
- shaking or tremors
- confusion
- sweating
- headache
- diarrhoea.
These are likely to occur in the first few days of stopping treatment or very rarely if you miss a dose. However, they are more likely to occur if you stop taking AROPAX too quickly.
Therefore, always consult your doctor before stopping your medicine. For the majority of patients, symptoms go away on their own within a few weeks.
However, if you feel that the unwanted symptoms are too severe, see your doctor who will suggest how to manage stopping treatment more slowly.
Although AROPAX is not recommended for children under 18 years of age, the most common unwanted effects in children under 18 are:
- decreased appetite
- tremor (uncontrollable trembling)
- sweating
- hyperactivity
- hostile/unfriendly behaviour
- agitation
- changing emotions, including crying, changes in mood, trying to harm themselves, thoughts of suicide and attempting suicide.
Additional symptoms that have been experienced by children whilst stopping treatment are changing emotions (including thoughts of suicide, attempting suicide, mood changes and feeling tearful), abdominal pain and nervousness.
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What AROPAX contains
| Active ingredient (main ingredient) | Paroxetine (as hydrochloride hemihydrate) |
| Other ingredients (inactive ingredients) |
|
Do not take this medicine if you are allergic to any of these ingredients.
AROPAX tablets do not contain sucrose, lactose or tartrazine.
What AROPAX looks like
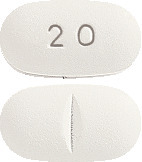
AROPAX tablets are white, film-coated, oval shaped biconvex tablet, debossed with "20" on one side and a break bar on the other containing 20 mg of paroxetine.
Available in blister packs of 30 tablets.
(Aust R 57927)
Who distributes AROPAX
Aspen Pharmacare Australia Pty Ltd
34-36 Chandos Street
St Leonards NSW 2065
Australia.
AROPAX® is a registered trademark of Aspen Global Incorporated.
This leaflet was prepared in September 2023.
Published by MIMS October 2023


 Paroxetine hydrochloride hemihydrate is an odourless, off white powder, with a melting point range of 120°C to 138°C and solubility of 5.4 mg/mL in water.
Paroxetine hydrochloride hemihydrate is an odourless, off white powder, with a melting point range of 120°C to 138°C and solubility of 5.4 mg/mL in water.