WHAT IS IN THIS LEAFLET
This leaflet answers some common questions about Bonefos.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Bonefos against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
WHAT BONEFOS IS USED FOR
Certain cancers increase bone breakdown and lead to an increased amount of calcium circulating in the bloodstream. This can result in high calcium levels in the blood (hypercalcaemia) or bone destruction (osteolysis) which increases your risk of bone fractures and bone pain.
Bonefos works by stopping calcium from coming out of your bones and keeps your blood calcium at normal levels (normocalcaemia). Bonefos can be used to either bring your blood calcium levels down to a normal level (acute treatment) or to keep it down at a normal level (maintenance treatment).
Bonefos can also be used to treat bone destruction due to breast cancer and bone marrow cancer.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is available only with a doctor’s prescription.
There is not enough information to recommend the use of this medicine for children.
BEFORE YOU TAKE BONEFOS
When you must not take it
Do not take Bonefos if you have an allergy to:
- sodium clodronate, the active ingredient in Bonefos
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not take Bonefos if you have or have had any of the following medical conditions:
- severe inflammation of the digestive system
Do not take Bonefos if you are taking any other ‘bisphosphonate’ medicine, a class of medicines that prevent the loss of bone mass (used in the treatment of osteoporosis and similar diseases) and is also used in the treatment of high calcium levels in the blood and/or bone destruction due to certain cancers.
Do not take this medicine after the expiry date printed on the carton and blister. The expiry date is printed on the carton and on each blister after “EXP” (e.g. 11 18 refers to November 2018). The expiry date refers to the last day of that month. If it has expired return it to your pharmacist for disposal.
Do not take this medicine if the packaging is torn or shows signs of tampering. If the packaging is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any medicines, foods, preservatives or dyes.
Tell your doctor if you:
- have or have had kidney disease
- are on a low sodium diet
- are having any invasive dental procedures e.g. tooth extractions
- any infections in the mouth
Tell your doctor if you need any dental work to be done. If you develop a rare side-effect affecting the jaw, dental work may make this condition worse.
Talk to your dentist about the need for any dental work to be done before starting Bonefos.
Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding. Your doctor can discuss with you the risks and benefits involved.
If you have not told your doctor about any of the above, tell him/her before you start taking Bonefos.
Taking other medicines
Tell your doctor or pharmacist if you are taking any medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and Bonefos may interfere with each other. These include:
- estamustine phosphate, an anti-cancer drug
- antacids, medicines used to treat heartburn and indigestion
- non steroidal anti-inflammatory drugs (NSAIDs), used to treat acute or chronic pain and conditions such as arthritis
- aminoglycoside antibiotics, given by injection, used to treat serious infections
- medicines that reduce your blood calcium levels [e.g. corticosteroids, phosphate, calcitonin, mithramycin and diuretics (medicines that increase the excretion of water from your body, and used to treat heart failure, liver cirrhosis, hypertension and certain kidney diseases)]
- iron supplements
These medicines may be affected by Bonefos or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
HOW TO TAKE BONEFOS
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions printed on the pharmacist label, ask your doctor or pharmacist for help.
How much to take
The following doses are likely to be recommended by your doctor if your kidneys function properly. If your kidneys do not function properly, your doctor may recommend a different dosage regimen.
Follow all directions given to you by your doctor or pharmacist carefully.
Treatment to initially bring blood calcium levels down to normal (acute treatment):
The usual starting dose of Bonefos is 2400-3200 mg in divided doses per day (e.g. three to four 800 mg tablets), depending on how quickly your calcium level drops.
Treatment to maintain blood calcium at normal levels:
The usual dose of Bonefos is 1600 mg in divided doses per day (e.g. four 400 mg capsules or two 800 mg tablets).
Treatment of bone destruction (osteolysis):
The usual dose of Bonefos is 1600 mg in divided doses per day (e.g. four 400 mg capsules or two 800 mg tablets).
How to take it
Swallow the medicine whole with a full glass of water. Do not crush or dissolve the medicine in water. The Bonefos 800 mg tablet may be broken in half to make swallowing easier but the halves have to be taken at the same time.
Do not chew the medicine.
Do not take the medicine with milk, food or medicines containing calcium.
When to take it
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
If possible take your medicine first thing in the morning, on an empty stomach. However, if you cannot take your medicine in the morning, or if your doctor has told you to split your dose, take your medicine at least 2 hours after food or drink (other than plain water). Certain medicines and food (particularly high calcium food such as milk and cheese) can interfere with the absorption of Bonefos. This may stop your body taking in as much medicine as it should, and then it may not work properly.
Do not eat or drink for an hour after taking Bonefos.
If you need to take an antacid, take it at least 2 hours before or 2 hours after your dose of Bonefos.
How long to take it
Continue taking your medicine for as long as your doctor tells you. This medicine helps to control your condition, but does not cure it. It is important to keep taking your medicine even if you feel well.
If you forget to take it
If you forget to take a dose of Bonefos, skip the dose you missed and take your next dose when you are meant to.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much Bonefos. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
WHILE YOU ARE TAKING BONEFOS
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Bonefos.
Tell any doctors, dentists, oral surgeons and pharmacists who treat you that you are taking this medicine.
Avoid invasive dental procedures whilst being treated with this medicine if possible. If you develop a rare side-effect affecting the jaw, dental work may make this condition worse.
Ensure you maintain good oral hygiene whilst taking this medicine. If you cannot avoid dental work whilst taking this medicine, oral hygiene is important to minimise the risk of developing side-effects.
Tell your doctor if you feel any thigh, hip or groin pain while taking Bonefos. These symptoms could be an early indication of a possible fracture.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine. It may affect other medicines used during surgery.
If you become pregnant while taking this medicine, tell your doctor immediately.
Drink plenty of fluid during treatment, particularly if you have high calcium levels in the blood or kidney disease.
Keep all of your doctor’s appointments so that your progress can be checked and your kidney function can be monitored. Your doctor may do some tests from time to time to make sure the medicine is working and to prevent unwanted side effects.
Things you must not do
Do not take Bonefos to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor. If you stop taking it suddenly, your condition may worsen or you may have unwanted side effects such as loss of appetite, nausea, vomiting, constipation and stomach pain (symptoms of hypercalcaemia). If possible, your doctor will gradually reduce the amount you take each day before stopping the medicine completely.
Do not take half a Bonefos 800 mg tablet as a substitute for one Bonefos 400 mg capsule.
SIDE EFFECTS
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Bonefos. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- nausea, vomiting or diarrhoea
- stomach pain
The above list includes the more common side effects of your medicine. They are usually mild and short-lived.
Tell your doctor, dentist or oral surgeon as soon as possible if you notice jaw-bone problems, which may include infection and delayed healing after teeth are pulled out or other work that involves drilling into the jaw. This may be a serious side effect that requires medical attention. Serious side effects are rare.
Tell your doctor as soon as possible if you notice any of the following:
- unusual bleeding or bruising under the skin, purple brown spots visible through skin
- difficulty breathing
- fever
- redness of the skin
- muscle spasm or twitching, numbness or tingling in fingers and toes, depression, irritability, confusion, disorientation (symptoms of low calcium levels in the blood)
- pain, weakness or discomfort in your thigh, hip or groin (this may be an early sign of a possible fracture of the thigh bone)
The above list includes serious side effects that may require medical attention. Serious side effects are rare.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell. Other side effects not listed above may also occur in some people.
AFTER TAKING BONEFOS
Storage
Keep your medicine in the pack until it is time to take them. If you take the tablets/capsules out of the pack they may not keep well.
Keep your medicine in a cool dry place where the temperature stays below 25°C.
Do not store Bonefos or any other medicine in the bathroom, near a sink, or on a window-sill.
Do not leave it in the car. Heat and damp can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Return any unused medicine to your pharmacist.
PRODUCT DESCRIPTION
What it looks like
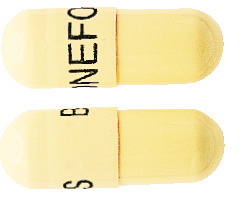
Bonefos capsules: pale yellow in colour with “BONEFOS” printed in black.
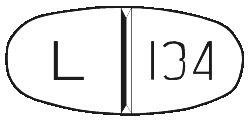
Bonefos tablets: white, oval shaped, scored and embossed with “L134” on one side.
Ingredients
Active ingredients per capsule:
- Bonefos 400 mg capsule – 400 mg of sodium clodronate.
Inactive ingredients per capsule:
- lactose
- purified talc
- silica colloidal anhydrous
- calcium stearate
- gelatin
- titanium dioxide
- iron oxide red
- iron oxide yellow
- purified water
- Tekprint SW-9008 black ink.
Active ingredient per tablet:
- Bonefos 800 mg tablet - 800 mg of sodium clodronate.
Inactive ingredients per tablet:
- croscarmellose sodium
- microcrystalline cellulose
- stearic acid
- magnesium stearate
- silica colloidal anhydrous
- Opadry II complete film coating system 85F18422 white.
Supplier
Made in Finland for:
Bayer Australia Limited
ABN 22 000 138 714
875 Pacific Highway
Pymble, NSW 2073
Australian Registration Numbers
- 400 mg capsules:
AUST R 66703 and 66704 - 800 mg tablets:
AUST R 181921 and 181922
Date of preparation
August 2013
See TGA website (www.ebs.tga.gov.au) for latest Australian Consumer Medicine Information.
® Registered Trademark of Bayer AG, Germany
©Bayer Australia Ltd
All rights reserved.

Published by MIMS November 2013

 A log-rank comparison of the proportion of patients remaining event free of nonvertebral fracture or hypercalcaemia over four years favoured the clodronate group (p = 0.021).
A log-rank comparison of the proportion of patients remaining event free of nonvertebral fracture or hypercalcaemia over four years favoured the clodronate group (p = 0.021).
 In the second trial, 144 women received either 1600 mg (2 x 400 mg capsules twice daily) of sodium clodronate (n = 73) or placebo (n = 71). Patients were followed for a period of one year. The log-rank comparison of the time patients remained free of a new bone event was marginally significant in favour of the clodronate group. Bone pain was significantly reduced in the clodronate group (see Table 4).
In the second trial, 144 women received either 1600 mg (2 x 400 mg capsules twice daily) of sodium clodronate (n = 73) or placebo (n = 71). Patients were followed for a period of one year. The log-rank comparison of the time patients remained free of a new bone event was marginally significant in favour of the clodronate group. Bone pain was significantly reduced in the clodronate group (see Table 4). In the multiple myeloma and breast cancer trials, there were substantial numbers of patients lost to follow-up, however this did not appear to advantage any particular treatment group.
In the multiple myeloma and breast cancer trials, there were substantial numbers of patients lost to follow-up, however this did not appear to advantage any particular treatment group.