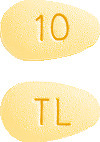

SUMMARY CMI
BRINTELLIX®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I using Brintellix?
Brintellix contains the active ingredient vortioxetine. Brintellix is used to treat major depression in adults.
For more information, see Section 1. Why am I using Brintellix? in the full CMI.
2. What should I know before I use Brintellix?
Do not use if you have ever had an allergic reaction to vortioxetine or any of the ingredients listed at the end of the CMI.
Talk to your doctor or pharmacist if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use Brintellix? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with Brintellix and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use Brintellix?
- Follow all directions given to you by your doctor or pharmacist carefully.
- Swallow the tablets whole with a full glass of water
More instructions can be found in Section 4. How do I use Brintellix? in the full CMI.
5. What should I know while using Brintellix?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Drinking alcohol |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using Brintellix? in the full CMI.
6. Are there any side effects?
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Brintellix.
Brintellix helps most people with depression, but it may have unwanted side effects in some people.
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
FULL CMI
BRINTELLIX®
Active ingredient: Vortioxetine hydrobromide (vor-tee-ox-e-teen high-dro-bro-mide)
Consumer Medicine Information (CMI)
This leaflet provides important information about using Brintellix. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using Brintellix.
Where to find information in this leaflet:
1. Why am I using Brintellix?
2. What should I know before I use Brintellix?
3. What if I am taking other medicines?
4. How do I use Brintellix?
5. What should I know while using Brintellix?
6. Are there any side effects?
7. Product details
1. Why am I using Brintellix?
Brintellix contains the active ingredient vortioxetine. Brintellix is thought to work by its actions on multiple brain chemicals including serotonin, noradrenaline, dopamine, histamine and acetylcholine which are thought to be involved in controlling mood and related mental processes.
Brintellix is used to treat major depression in adults. Depression is longer lasting or more severe than the "low moods" everyone has from time to time due to the stress of everyday life. It is thought to be caused by a chemical imbalance in parts of the brain. This imbalance affects your whole body and can cause emotional, physical and cognitive symptoms such as feeling low in spirit, reduced ability to think or concentrate, or indecisiveness, loss of interest in activities, being unable to enjoy life, poor appetite or overeating, disturbed sleep, often waking up early, loss of sex drive, lack of energy and feeling guilty over nothing.
2. What should I know before I use Brintellix?
Warnings
Do not use Brintellix if:
- you are allergic to vortioxetine, or any of the ingredients listed at the end of this leaflet.
Always check the ingredients to make sure you can use this medicine.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not take Brintellix at the same time as the following other medicines:
- monoamine oxidase inhibitors (MAOIs), such as phenelzine, tranylcypromine, selegiline, rasagiline and moclobemide
- medicines which act like a monoamine oxidase inhibitor, such as the antibiotic linezolid
Taking Brintellix with MAOIs may cause a serious reaction including sudden changes in mental state, twitching, rapid heartbeat, high blood pressure, fever and diarrhoea.
If you have taken an MAOI, you will need to wait at least 14 days before starting Brintellix. If you have used moclobemide, one day must elapse after you stop taking moclobemide before you start taking Brintellix.
After stopping Brintellix, you must allow at least 14 days before taking any MAOI or moclobemide.
Do not take Brintellix after the expiry date (EXP) printed on the pack. The expiry date refers to the last day of the month.
It may have no effect at all or, an entirely unexpected effect if you take it after the expiry date.
Do not take Brintellix if the packaging is torn or shows signs of having been tampered with.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Check with your doctor or pharmacist if you:
- have any of the following medical conditions:
- A tendency to bleed or bruise easily, or if you are pregnant, as there may be an increased risk of heavy vaginal bleeding shortly after birth, especially if you have a history of bleeding disorders.
- Low sodium levels in the blood
- Seizures or fits
- A history of suicide-related events or suicidal ideas
- A history, or family history, of mania or bipolar disorder (manic depression)
- Other diseases affecting the brain, including psychiatric conditions
- Other significant medical illnesses, such as unstable heart disease, stroke or severe liver or kidney disease
- Glaucoma, risk of glaucoma, or any condition affecting the fluid pressure in the eye. If your eyes become painful and you develop blurred vision during treatment, contact your doctor - take any medicines for any other condition
- When you are on antidepressant treatment, including vortioxetine, you may also experience feelings of aggression, agitation, anger and irritability. If this occurs, you should talk to your doctor.
- Tell your doctor if you are 65 years of age or older.
- Tell your doctor if you are receiving electroconvulsive therapy.
- Do not give Brintellix to a child or adolescent.
- Brintellix is not recommended in children or adolescents less than 18 years of age.
- If you have not told your doctor about any of the above, tell them before you use Brintellix.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
Brintellix should not be used during pregnancy unless the benefit outweighs the risk. Your doctor can discuss with you the risks and benefits involved. Brintellix may increase the risk of postpartum haemorrhage.
When taken during pregnancy, particularly in the last three months of pregnancy, medicines like Brintellix may increase the risk of a serious condition in babies called persistent pulmonary hypertension of the newborn (PPHN), making the baby breathe faster and appear bluish. These symptoms usually begin during the first 24 hours after the baby is born.
Other effects on your baby may include difficulty breathing, fits, body temperature changes, feeding difficulties, vomiting, stiff or floppy muscles, tremor, jitteriness, irritability, lethargy, constant crying, sleepiness or sleeping difficulties.
If your newborn baby has any of the above symptoms, you should contact your doctor immediately.
It is recommended that you do not breast-feed while taking Brintellix, as it may be excreted in the milk.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with Brintellix and affect how it works. These include:
- Monoamine oxidase inhibitors (MAOIs), including selegiline, rasagiline, linezolid and moclobemide.
You must stop taking MAOIs at least two weeks before starting Brintellix. You must wait at least one day after finishing moclobemide before you start Brintellix.
You must stop taking Brintellix at least two weeks before you start taking an MAOI or moclobemide.
- rifampicin, an antibiotic
- St. John's Wort, a herbal remedy
- sumatriptan (and similar medicines ending in 'triptan'), medicines used to treat migraine headaches
- lithium, used to treat mood swings and some types of depression
- tryptophan, an amino acid
- tramadol and other similar medicines (opioid based pain killers) used to relieve pain, these are known to increase the risk of a rare condition called serotonin syndrome
- mefloquine, an anti-malaria medicine
- bupropion, a medicine for nicotine dependence
- medicines known to cause low sodium level in the blood
- medicines used to thin the blood or known to prolong bleeding e.g. warfarin, dipyridamole, aspirin, and non-steroidal anti-inflammatory drugs (NSAIDs)
- antipsychotics, a class of medicines used to treat mental illness, e.g. risperidone, olanzapine, quetiapine
- tricyclic antidepressants, e.g. imipramine, desipramine
- any other medicines for depression, anxiety, panic disorder, obsessive-compulsive disorder or pre-menstrual dysphoric disorder
These medicines may be affected by Brintellix, or may affect how well it works. You may need to use different amounts of medicines, or take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
If you have not told your doctor or pharmacist about any of these things, tell them before you take Brintellix.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect Brintellix.
If you are having a urine drug screen, taking Brintellix may cause positive results for methadone when some test methods are used, even though you may not be taking methadone. If this happens, a more specific test can be performed.
4. How do I use Brintellix?
Follow all directions given to you by your doctor or pharmacist carefully.
They may differ from the information contained in this leaflet.
If you do not understand the instructions on the label, ask your doctor or pharmacist for help.
How much to take
- Your doctor will tell you how much Brintellix to take each day. Take the amount your doctor tells you to.
- The usual dose for Brintellix in adults less than 65 years of age is 10 mg taken orally as one daily dose. The dose may be increased by your doctor to a maximum of 20 mg per day or lowered to a minimum of 5 mg per day.
- For elderly people 65 years of age or older, the starting dose is 5 mg taken once daily.
- Ask your doctor or pharmacist if you are unsure of the correct dose for you.
- They will tell you exactly how much to take.
- Follow the instructions they give you.
- If you take the wrong dose, Brintellix may not work as well and your condition may not improve.
When to take Brintellix
- Take Brintellix as a single dose either in the morning or in the evening.
- Take your medicine at about the same time each day.
- It does not matter if you take Brintellix before or after food.
How to take Brintellix
- Swallow the tablets whole with a full glass of water.
How long to take it
Continue to take Brintellix even if it takes some time before you feel any improvement in your condition.
It may take two weeks, sometimes longer, before you feel any improvement.
Continue to take Brintellix for as long as your doctor recommends.
If your depression resolves while taking BRINTELLIX treatment should be continued for at least 6 months after you feel well again.
Your doctor will check your progress at regular intervals.
Do not change the dose or stop taking this medicine without first checking with your doctor.
If you forget to use Brintellix
Take the next dose at the usual time.
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Do not take a double dose to make up for the dose that you missed.
This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you use too much Brintellix
If you think that you have used too much Brintellix, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26 (Australia) or 0800 764 766 (New Zealand)), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
Keep the telephone number handy.
Some of the signs of an overdose could be dizziness, feeling sick (nausea), diarrhoea, stomach discomfort, itching on the whole body, sleepiness and flushing.
Following intake of dosage several times higher than the prescribed dose, fits (seizures) and a rare condition called serotonin syndrome have been reported.
5. What should I know while using Brintellix?
Things you should do
Tell your doctor immediately if you become pregnant while taking Brintellix.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Brintellix.
Remind any doctor, dentist or pharmacist you visit that you are using Brintellix.
Keep all of your doctor's appointments so that your progress can be checked.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking Brintellix.
It may affect other medicines used during surgery.
If you have thoughts about killing yourself or other mental/mood changes, contact your doctor immediately or go to the nearest hospital for treatment.
All mentions of suicide or violence must be taken seriously.
Occasionally, the symptoms of depression may include thoughts of suicide or self-harm. It is possible that these symptoms continue or get worse until the full antidepressant effect of the medicine becomes apparent.
This is more likely to occur:
- if you are a young adult, i.e. 18 to 24 years of age; or
- you have previously had thoughts about killing or harming yourself
You may find it helpful to tell a relative or close friend that you are depressed and ask them to read this leaflet. You might ask them to tell you if they think your depression is getting worse, or if they are worried about changes in your behaviour.
Patients and care givers should pay attention for any of the following warning signs of suicide-related behaviour while taking Brintellix:
- worsening of depression
- thoughts or talk of death or suicide
- thoughts or talk of self-harm or harm to others
- any recent attempts of self-harm
- increase in aggressive behaviour, irritability, agitation or any other unusual changes in behaviour or mood.
Things you should not do
Do not take Brintellix to treat any other complaints unless your doctor tells you to.
Do not give Brintellix to anyone else, even if they have the same condition as you.
Do not stop taking Brintellix, or lower the dosage, without checking with your doctor.
If you stop taking it suddenly, or reduce the amount you take, your condition may worsen.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how Brintellix affects you.
Drinking alcohol
Tell your doctor or pharmacist if you drink alcohol.
Avoid alcohol while you are taking Brintellix.
Looking after your medicine
Follow the instructions in the carton on how to take care of your medicine properly.
Keep Brintellix in the blister pack until it is time to take them.
If you take the tablets out of the box or blister pack they may not keep well.
Store it in a cool dry place below 30°C away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Heat and dampness can destroy some medicines.
Keep it where young children cannot reach it.
A locked cupboard at least one-and-a-half metres above ground is a good place to store medicines.
Getting rid of any unwanted medicine
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine you may have left over.
Do not use this medicine after the expiry date.
6. Are there any side effects?
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Brintellix.
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions.
Side effects are generally mild to moderate and occur within the first two weeks of treatment. They are usually temporary and generally do not require treatment cessation.
Less serious/common side effects
| Less serious/common side effects | What to do |
| Speak to your doctor or pharmacist if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
| Call your doctor straight away if you notice any of these serious side effects. |
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
* The side effects marked with an asterisk (*) are a number of rare side effects that are known to occur with medicines that work in a similar way to Brintellix.
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
7. Product details
What Brintellix contains
| Active ingredient (main ingredient) |
|
| Other ingredients (inactive ingredients) |
|
| Potential allergens | Brintellix does not contain lactose, gluten, sucrose, tartrazine or any other azo dyes. |
Do not take this medicine if you are allergic to any of these ingredients.
What Brintellix looks like
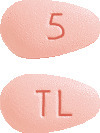
- Brintellix 5 mg tablets are pink, almond-shaped, biconvex film-coated tablet engraved with "TL" on one side and "5" on the other side.
- Brintellix 10 mg tablets are yellow, almond-shaped, biconvex film-coated tablet engraved with "TL" on one side and "10" on the other side.
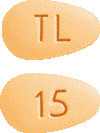
- Brintellix 15 mg tablets are orange, almond-shaped, biconvex film-coated tablet engraved with "TL" on one side and "15" on the other side.
- Brintellix 20 mg tablets are red, almond-shaped, biconvex film-coated tablet engraved with "TL" on one side and "20" on the other side.
Brintellix is available in blister packs of 28 tablets.
Australian Registration Numbers are:
5 mg tablet: AUST R 203986
10 mg tablet: AUST R 203955
15 mg tablet: AUST R 203981
20 mg tablet: AUST R 203970
New Zealand Registration Numbers are:
5 mg tablet: TT50-10588
10 mg tablet: TT50-10588a
15 mg tablet: TT50-10588b
20 mg tablet: TT50-10588c
Who distributes Brintellix
Brintellix is made by H. Lundbeck A/S, Denmark.
Distributed in Australia by:
Lundbeck Australia Pty Ltd
1 Innovation Road
North Ryde NSW 2113
Ph: 02 8669 1000
Distributed in New Zealand by:
Pharmacy Retailing t/a Healthcare Logistics
58 Richard Pearse Drive
Mangere, Auckland 2022
Ph: 0800 540 555
This leaflet was prepared in March 2022
Brintellix is a registered trademark of H. Lundbeck A/S.
Published by MIMS May 2022

 The effect of vortioxetine on sexual function was further evaluated in an 8-week, double-blind, flexible-dose, comparative study (n=424) versus escitalopram in patients treated for at least 6 weeks with an SSRI (citalopram, paroxetine, or sertraline), with a low level of depressive symptoms (baseline CGI-S ≤ 3) and TESD induced by the prior SSRI treatment. Vortioxetine 10-20 mg/day had statistically significantly less TESD than escitalopram 10-20 mg/day as measured by change in the CSFQ-14 total score (2.2 points, p=0.013) at week 8. The proportion of responders was not significantly different in the vortioxetine group (162 (74.7%)) compared with the escitalopram group (137 (66.2%)) at week 8 (OR 1.5 p=0.057). The antidepressant effect was maintained in both treatment groups. See Figure 1.
The effect of vortioxetine on sexual function was further evaluated in an 8-week, double-blind, flexible-dose, comparative study (n=424) versus escitalopram in patients treated for at least 6 weeks with an SSRI (citalopram, paroxetine, or sertraline), with a low level of depressive symptoms (baseline CGI-S ≤ 3) and TESD induced by the prior SSRI treatment. Vortioxetine 10-20 mg/day had statistically significantly less TESD than escitalopram 10-20 mg/day as measured by change in the CSFQ-14 total score (2.2 points, p=0.013) at week 8. The proportion of responders was not significantly different in the vortioxetine group (162 (74.7%)) compared with the escitalopram group (137 (66.2%)) at week 8 (OR 1.5 p=0.057). The antidepressant effect was maintained in both treatment groups. See Figure 1. In a 5-week, double-blind, placebo- and paroxetine-controlled study in healthy subjects with normal sexual functioning, without the confounding effect of depression, vortioxetine 10 mg/day (2.74 points, p=0.009) was statistically significantly better than paroxetine 20 mg/day as measured by change in the CSFQ-14 total score, whereas vortioxetine 20 mg/day was not (1.05 points, p=0.303). Neither vortioxetine 10 mg/day nor 20 mg/day were significantly worse than placebo. See Figure 2.
In a 5-week, double-blind, placebo- and paroxetine-controlled study in healthy subjects with normal sexual functioning, without the confounding effect of depression, vortioxetine 10 mg/day (2.74 points, p=0.009) was statistically significantly better than paroxetine 20 mg/day as measured by change in the CSFQ-14 total score, whereas vortioxetine 20 mg/day was not (1.05 points, p=0.303). Neither vortioxetine 10 mg/day nor 20 mg/day were significantly worse than placebo. See Figure 2.




