What is in this leaflet
This leaflet answers some common questions about this medicine. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking this medicine against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What this medicine is used for
The name of your medicine is Capecitabine APOTEX. It contains the active ingredient capecitabine.
It is used to treat cancer of the:
- bowel and rectum (colorectal)
- breast and stomach and food pipe (oesophagus).
Capecitabine belongs to a group of medicines called anti-neoplastic agents. Within this group, capecitabine belongs to a class of medicines called fluoropyrimidine analogues.
Capecitabine is converted by the liver and cancer cells to another medicine called 5-fluorouracil (also called 5-FU).
It is 5-FU that acts to kill or stop the growth of cancer cells.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed this medicine for another reason.
There is no evidence that this medicine is addictive.
This medicine is available only with a doctor's prescription.
Do not give capecitabine to children. Safety and effectiveness of this medicine in persons under 18 years of age have not been established.
Before you take this medicine
When you must not use it
Do not take this medicine if you have an allergy to:
- capecitabine
- 5-fluorouracil (also called 5-FU)
- other fluoropyrimidine medicines
- any of the ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not take this medicine if you have any of the following medical conditions:
- severe kidney disease
- dihydropyramidine dehydrogenase (DPD) deficiency
Do not take this medicine if you are taking a medicine containing sorivudine or related medicines such as brivudine. Taking these medicines at the same time as capecitabine is potentially fatal.
Do not take this medicine if you are pregnant. Capecitabine may be harmful to an unborn baby when taken by a pregnant woman. It is not recommended that you take capecitabine while you are pregnant. Additionally, if you are a woman, you should use effective contraception to avoid becoming pregnant while you are taking capecitabine.
Do not breastfeed if you are taking this medicine. It is not known whether capecitabine and 5-FU pass into breast milk and there is a possibility that your baby may be affected.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- heart disease
- liver disease
- kidney disease
- dehydration
Some signs and symptoms of dehydration include:
- dry skin
- dark coloured urine
- thirst
- weakness or fatigue
- loss of appetite
Tell your doctor if you are pregnant or plan to become pregnant or are breastfeeding.
If you have not told your doctor about any of the above, tell them before you start taking this medicine.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and capecitabine may interfere with each other. These include:
- medicines used to thin the blood such as warfarin and phenprocoumon
- phenytoin, used to treat epilepsy and heart irregularities
- leucovorin, also called folinic acid, used to treat folic acid deficient anaemias
- antacids, used to treat heart burn or indigestion
These medicines may be affected by capecitabine or may affect the way it works. You may need different amounts of your medicines or may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take this medicine
Follow carefully all directions given to you by your doctor carefully. Their instructions may be different to the information in this leaflet.
If you are not sure how to take this medicine, ask your doctor or pharmacist for help.
How much to take
Take this medicine exactly as your doctor has prescribed.
Capecitabine may be given with or without chemotherapy.
Your doctor will tell you how much of capecitabine you should take. This will depend on your condition, whether you are taking any other medicines and your height and weight. Elderly patients may be prescribed a lower dose.
Your doctor may want you to take a combination of 150 mg (light peach colour) and 500 mg (peach colour) tablets for each dose.
If a combination of tablets is prescribed, it is very important that you correctly identify the tablets.
How to take it
Swallow the tablets whole with a glass of water.
Do not chew the tablets.
When to take it
Capecitabine tablets should be taken with food. You should take this medicine no later than 30 minutes after food.
Take this medicine at the same time each day. Taking it at the same time each day will have the best effect and will also help you remember when to take it.
When taken in combination with chemotherapy, your doctor will advise which days of your treatment cycle capecitabine should be taken.
If you are not sure when to take capecitabine, ask your doctor.
How long to take it
The duration of treatment with capecitabine varies, depending on the nature of your illness and your individual response to the treatment.
Capecitabine therapy is made up of a series of treatment cycles which usually lasts for 21 days.
Your doctor will advise you how many cycles of treatment you will have and whether there are any rest days in the cycle.
In most cases, your treatment cycle will consist of intermittent capecitabine therapy, where you will take capecitabine for 14 days, followed by a rest period of 7 days.
During the rest period, you will not take any capecitabine.
Alternatively, your treatment cycle may be continuous, which involves 21 days of capecitabine treatment and no rest period.
Continue taking your medicine for as long as your doctor tells you.
If you forget to take it
Skip the missed dose and take your next dose at the usual time.
Do not take a double dose to make up for missed doses. This may increase the chance of you experiencing side effects.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints to help you remember.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much of this medicine. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are taking this medicine
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking this medicine.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine. It may affect other medicines used during surgery.
If you become pregnant or start to breastfeed while taking this medicine, tell your doctor immediately.
If you are about to have any blood tests, tell your doctor that you are taking this medicine. It may interfere with the results of some tests.
Keep all your doctor's appointments so that your progress can be checked. Your doctor may do some tests from time to time to make sure the medicine is working and to prevent unwanted side effects.
Take this medicine exactly as your doctor has told you to.
Tell your doctor if, for any reason, you have not taken your medicine exactly as prescribed.
Tell your doctor if you feel the tablets are not helping your condition.
Things you must not do
Do not take this medicine to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
Do not take any other medicines whether they require a prescription or not without first telling your doctor or consulting with a pharmacist.
Things to be careful of
Be careful driving or operating machinery until you know how capecitabine affects you.
Side effects
Tell your doctor as soon as possible if you do not feel well while you are taking this medicine or if you have any questions or concerns.
All medicines can have side effects. Sometimes they are serious but most of the time they are not.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Tell your doctor if you notice any of the following:
- diarrhoea
- vomiting
- nausea (feeling like you want to vomit)
- fatigue (tiredness), weakness or weariness
- skin rashes, discolouration, dry or itchy skin
- abdominal (stomach) pain
- fever, or increased temperature sensitivity
- constipation
- headache
- dizziness
- loss of appetite, weight loss
- hair loss
- increased eye watering or irritation, sensitivity to light, conjunctivitis (itchy eyes and crusty eyelids)
- taste disturbance
- indigestion, wind
- dry mouth, thirst
- sore mouth, mouth ulcers, cold sores
- nail disorders
- sore throat, cough, nose bleeds
- shortness of breath, difficulty in breathing, or tightening of the chest
- redness or swelling of your hands and/or feet
- tingling or numbness of the hands or feet
- back pain, muscle and joint pain
- dark coloured urine
- difficulty sleeping
- infections
- depression
Tell your doctor immediately if you notice any of the following:
- severe diarrhoea with more than 4 bowel movements each day
- nausea that has reduced your appetite significantly
- vomiting more than once in a 24-hour period
- pain, redness and/or swelling of your hands and/or feet that has affected your normal activities (hand-foot-syndrome)
- pain, redness, swelling or ulcers in the mouth (stomatitis)
- passing little or no urine (this could be kidney disease) – other symptoms include drowsiness, nausea, vomiting and breathlessness
You need to stop taking capecitabine if you experience the above side effects. Your doctor will treat your side effects before they start you on capecitabine again.
Tell your doctor immediately or go to your nearest Accident and Emergency if you notice any of the following:
- chest pain
- irregular heart beat
- shortness of breath
- confusion, disorientation or memory loss
- poor balance or lack of coordination
- decreased strength or progressive weakness in your body, numbness or weakness of arms or legs
- blurred or loss of vision
- signs of infection such as swelling, redness and increased temperature
- signs of liver disease such as yellowing of the skin and eyes
- blood in the faeces
- severe blisters and bleeding in the lips, eyes, mouth, nose and genitals
- severe skin reaction which starts with painful red areas, then large blisters and ends with peeling of layers of skin – this is accompanied by fever and chills, aching muscles and generally feeling unwell
- symptoms of an allergic reaction including cough, shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue, throat or other parts of the body; rash, itching or hives on the skin
These are serious side effects. You may need urgent medical attention.
These side effects may differ when taking capecitabine in combination with a chemotherapy medicine.
Please consult your doctor for possible side effects that may be caused by taking capecitabine with a chemotherapy medicine.
Other side effects not listed above may occur in some patients.
Tell your doctor if you notice anything else that is making you feel unwell, even if it is not on this list. Some of these side effects, for example changes in triglycerides, can only be found when your doctor does tests from time to time to check your progress.
Storage and disposal
Storage
Keep your tablets in the blister pack until it is time to take them. If you take the tablets out of the blister pack it will not keep well.
Keep your tablets in a cool dry place where the temperature will stay below 25°C.
Do not store your medicine, or any other medicine, in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep this medicine where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What Capecitabine APOTEX looks like
Capecitabine APOTEX 150 mg tablets: Light peach, oval, biconvex, film-coated tablet, engraved "APO" on one side, "C150" on the other side. AUST R 202726.
Blister packs of 60 tablets.
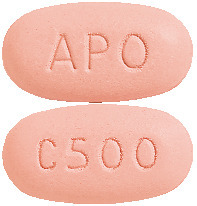
Capecitabine APOTEX 500 mg tablets: Peach, oval, biconvex, filmcoated tablet, engraved "APO" on one side, "C500" on the other side. AUST R 202738.
Blister packs of 120 tablets.
Not all strengths, pack types and/or pack sizes may be available.
Ingredients
Capecitabine APOTEX tablets contain either 150 mg or 500 mg of capecitabine as the active ingredient.
They also contain the following inactive ingredients:
- lactose
- croscarmellose sodium
- methylcellulose
- magnesium stearate
- colloidal anhydrous silica
- hypromellose
- hyprolose
- macrogol 8000
- titanium dioxide
- iron oxide red
- iron oxide yellow
This medicine is gluten-free, sucrose-free, tartrazine-free and free of other azo dyes.
Sponsor
Apotex Pty Ltd
16 Giffnock Avenue
Macquarie Park, NSW 2113
Australia
This leaflet was last updated in September 2019.
Published by MIMS November 2019


 Study M66001 did not include patients with Dukes' stage B disease. However, the findings of the study are considered to support the use of capecitabine as adjuvant therapy in patients with high risk stage B disease, such as those with inadequately sampled nodes, T4 lesions, perforation or poorly differentiated histology.
Study M66001 did not include patients with Dukes' stage B disease. However, the findings of the study are considered to support the use of capecitabine as adjuvant therapy in patients with high risk stage B disease, such as those with inadequately sampled nodes, T4 lesions, perforation or poorly differentiated histology. Capecitabine was equivalent to 5-FU/ leucovorin in time to disease progression, equivalent in overall survival and superior in objective response rate.
Capecitabine was equivalent to 5-FU/ leucovorin in time to disease progression, equivalent in overall survival and superior in objective response rate. Noninferiority of the capecitabine containing arms compared with the FOLFOX-4 containing arms in the overall comparison was demonstrated in terms of progression free survival (PFS) in the eligible per protocol population (EPP), with progression determined by the study investigators who were not blinded to treatment allocation (see Table 11). The criterion set for concluding noninferiority was that the upper limit of the 97.5% confidence interval for the hazard ratio for PFS was less than 1.23. The results for OS are similar to those reported for PFS. A comparison of capecitabine plus BV versus FOLFOX-4 plus BV was a prespecified exploratory analysis. In this treatment subgroup comparison, capecitabine plus BV was similar compared to FOLFOX-4 plus BV in terms of PFS (hazard ratio 1.01 [97.5% CI 0.84, 1.22]). The median follow up at the time of the primary analyses in the intent to treat population was 1.5 years; data from analyses following an additional 1 year of follow up are included in Table 12.
Noninferiority of the capecitabine containing arms compared with the FOLFOX-4 containing arms in the overall comparison was demonstrated in terms of progression free survival (PFS) in the eligible per protocol population (EPP), with progression determined by the study investigators who were not blinded to treatment allocation (see Table 11). The criterion set for concluding noninferiority was that the upper limit of the 97.5% confidence interval for the hazard ratio for PFS was less than 1.23. The results for OS are similar to those reported for PFS. A comparison of capecitabine plus BV versus FOLFOX-4 plus BV was a prespecified exploratory analysis. In this treatment subgroup comparison, capecitabine plus BV was similar compared to FOLFOX-4 plus BV in terms of PFS (hazard ratio 1.01 [97.5% CI 0.84, 1.22]). The median follow up at the time of the primary analyses in the intent to treat population was 1.5 years; data from analyses following an additional 1 year of follow up are included in Table 12. Study NO16966 also demonstrated superiority of the bevacizumab containing arms over placebo containing arms.
Study NO16966 also demonstrated superiority of the bevacizumab containing arms over placebo containing arms. Capecitabine was demonstrated to be noninferior to FOLFOX-4 in terms of PFS in the per protocol population (see Table 14). The criterion set for concluding noninferiority was the upper limit of the 95% confidence interval for the hazard ratio for PFS was less than 1.30. The results for overall survival were similar to those for PFS. The median follow up at the time of primary analyses in the intent to treat population was 2.1 years; data from analyses following an additional 6 months of follow up are also included in Table 14.
Capecitabine was demonstrated to be noninferior to FOLFOX-4 in terms of PFS in the per protocol population (see Table 14). The criterion set for concluding noninferiority was the upper limit of the 95% confidence interval for the hazard ratio for PFS was less than 1.30. The results for overall survival were similar to those for PFS. The median follow up at the time of primary analyses in the intent to treat population was 2.1 years; data from analyses following an additional 6 months of follow up are also included in Table 14. A pooled analysis of the efficacy data from first line (study NO16966; initial 2 arm part) and second line treatment (study NO16967) further support the noninferiority results of capecitabine versus FOLFOX-4 as obtained in the individual studies: PFS in the per protocol population (hazard ratio 1.00 [95% CI: 0.88; 1.14]) with a median PFS of 193 days (capecitabine; 508 patients) versus 204 days (FOLFOX-4; 500 patients). The results also indicate that capecitabine is comparable to FOLFOX-4 in terms of OS (hazard ratio 1.01 [95% CI: 0.87; 1.17]) with a median OS of 468 days (capecitabine) versus 478 days (FOLFOX-4).
A pooled analysis of the efficacy data from first line (study NO16966; initial 2 arm part) and second line treatment (study NO16967) further support the noninferiority results of capecitabine versus FOLFOX-4 as obtained in the individual studies: PFS in the per protocol population (hazard ratio 1.00 [95% CI: 0.88; 1.14]) with a median PFS of 193 days (capecitabine; 508 patients) versus 204 days (FOLFOX-4; 500 patients). The results also indicate that capecitabine is comparable to FOLFOX-4 in terms of OS (hazard ratio 1.01 [95% CI: 0.87; 1.17]) with a median OS of 468 days (capecitabine) versus 478 days (FOLFOX-4). Data from a randomised multicenter, phase III study comparing capecitabine to 5-FU and oxaliplatin to cisplatin in patients with previously untreated locally advanced or metastatic oesophagogastric cancer supports the use of capecitabine for the first line treatment of advanced oesophagogastric cancer (REAL-2). In this trial, 1002 patients were randomised in a 2 x 2 factorial design to one of the following 4 arms. See Table 16.
Data from a randomised multicenter, phase III study comparing capecitabine to 5-FU and oxaliplatin to cisplatin in patients with previously untreated locally advanced or metastatic oesophagogastric cancer supports the use of capecitabine for the first line treatment of advanced oesophagogastric cancer (REAL-2). In this trial, 1002 patients were randomised in a 2 x 2 factorial design to one of the following 4 arms. See Table 16. The primary efficacy analyses in the per protocol population demonstrated noninferiority in OS for capecitabine versus 5-FU-based regimens (hazard ratio 0.86, 95% CI: 0.80 to 0.99) and for oxaliplatin versus cisplatin based regimens (hazard ratio 0.92, 95% CI: 0.80 to 1.10). The median OS was 10.9 months in capecitabine based regimens and 9.6 months in 5-FU-based regimens. The median OS was 10.0 months in cisplatin based regimens and 10.4 months in oxaliplatin based regimens.
The primary efficacy analyses in the per protocol population demonstrated noninferiority in OS for capecitabine versus 5-FU-based regimens (hazard ratio 0.86, 95% CI: 0.80 to 0.99) and for oxaliplatin versus cisplatin based regimens (hazard ratio 0.92, 95% CI: 0.80 to 1.10). The median OS was 10.9 months in capecitabine based regimens and 9.6 months in 5-FU-based regimens. The median OS was 10.0 months in cisplatin based regimens and 10.4 months in oxaliplatin based regimens. A prospectively defined clinical benefit response score (pain, analgesic consumption and Karnofsky performance status) was used to assess the effect of treatment on tumour associated morbidity. The overall clinical benefit response was positive in 29 patients (20%) in the first trial and 8 patients (15%) in the second trial, 45 patients (31%) and 22 patients (41%), respectively, remained stable.
A prospectively defined clinical benefit response score (pain, analgesic consumption and Karnofsky performance status) was used to assess the effect of treatment on tumour associated morbidity. The overall clinical benefit response was positive in 29 patients (20%) in the first trial and 8 patients (15%) in the second trial, 45 patients (31%) and 22 patients (41%), respectively, remained stable.



 Amongst patients with colorectal cancer aged 60-79 years receiving capecitabine monotherapy in the metastatic setting, the incidence of grade 3 and 4 toxicity was similar to that in the overall population. In patients aged 80 years or older, a larger percentage experienced reversible grade 3 or 4 adverse reactions. When capecitabine was used in combination with other agents, elderly patients (> 65 years of age) experienced more grade 3 and 4 adverse reactions (ADRs) and ADRs that led to discontinuation than younger patients. An analysis of safety data in patients equal to or greater than 60 years of age treated with capecitabine in combination with docetaxel showed an increase in the incidence of treatment related grade 3 or 4 adverse reactions, treatment related serious adverse reactions and early withdrawals from treatment due to adverse reactions compared to patients less than 60 years of age.
Amongst patients with colorectal cancer aged 60-79 years receiving capecitabine monotherapy in the metastatic setting, the incidence of grade 3 and 4 toxicity was similar to that in the overall population. In patients aged 80 years or older, a larger percentage experienced reversible grade 3 or 4 adverse reactions. When capecitabine was used in combination with other agents, elderly patients (> 65 years of age) experienced more grade 3 and 4 adverse reactions (ADRs) and ADRs that led to discontinuation than younger patients. An analysis of safety data in patients equal to or greater than 60 years of age treated with capecitabine in combination with docetaxel showed an increase in the incidence of treatment related grade 3 or 4 adverse reactions, treatment related serious adverse reactions and early withdrawals from treatment due to adverse reactions compared to patients less than 60 years of age. Skin fissures were reported to be at least remotely related to capecitabine in less than 2% of the patients in seven completed clinical trials (n = 949).
Skin fissures were reported to be at least remotely related to capecitabine in less than 2% of the patients in seven completed clinical trials (n = 949). Hypersensitivity reactions (2%) and cardiac ischaemia/ infarction (3%) have been reported commonly for capecitabine in combination with other chemotherapy but in less than 5% of patients.
Hypersensitivity reactions (2%) and cardiac ischaemia/ infarction (3%) have been reported commonly for capecitabine in combination with other chemotherapy but in less than 5% of patients.




