What is in this leaflet
Read all of this leaflet carefully before you start taking this medicine.
This leaflet answers some of the common questions about EVIPLERA tablets. It does not contain all of the available information.
It does not take the place of talking to your doctor or pharmacist about your medical condition or treatment. If you have further questions, please ask your doctor or your pharmacist.
Keep this leaflet with your EVIPLERA medicine. You may need to read it again.
This medicine has been prescribed for you personally and you should not pass it on to others. It may harm them, even if their symptoms are the same as yours.
What is EVIPLERA
EVIPLERA is used to treat Human Immunodeficiency Virus (HIV-1) infection in adults.
EVIPLERA consists of three medicines:
- tenofovir disoproxil fumarate, also called tenofovir DF
- emtricitabine or FTC
- rilpivirine
These are combined in one tablet to help control Human Immunodeficiency Virus (HIV-1) infection.
Tenofovir disoproxil fumarate and emtricitabine belong to a group of antiviral medicines known as nucleoside and nucleotide reverse transcriptase inhibitors (NRTI).
Rilpivirine belongs to a group of antiviral medicines known as non-nucleoside reverse transcriptase inhibitors (NNRTI).
How EVIPLERA works
HIV-1 infection destroys CD4 T cells, which are important to the immune system. The immune system helps fight infection. After a large number of T cells are destroyed, acquired immune deficiency syndrome (AIDS) may develop.
EVIPLERA helps block HIV-1 reverse transcriptase, a viral chemical in your body (enzyme) that is needed for HIV-1 to multiply. EVIPLERA lowers the amount of HIV-1 in the blood (viral load). EVIPLERA may also help to increase the number of T cells (CD4+ cells), allowing your immune system to improve. Lowering the amount of HIV-1 in the blood lowers the chance of death or infections that happen when your immune system is weak (opportunistic infections).
Use in Children
EVIPLERA is for adults. EVIPLERA has not been studied in children under the age of 18 or adults over the age of 65.
Does EVIPLERA cure HIV OR AIDS
EVIPLERA does not cure HIV-1 infection or AIDS.
The long-term effects of EVIPLERA are not known at this time. People taking EVIPLERA may still get opportunistic infections or other conditions that happen with HIV-1 infection. Opportunistic infections are infections that develop because the immune system is weakened.
Some of these conditions are:
- pneumonia,
- herpes virus infections, and
- Mycobacterium avium complex (MAC) infection.
This medicine is only available from a pharmacist after it has been prescribed by a doctor who specialises in the treatment of HIV-1 infection.
If you wish to continue receiving treatment with EVIPLERA it is important you remain under the care of a hospital or doctor who specialises in the treatment of HIV-1 infection.
Does EVIPLERA reduce the risk of passing HIV to others
EVIPLERA does not stop you from transmitting HIV-1 to others.
For your health and the health of others, it is important to always practice safe sex by using a latex or polyurethane condom of other barrier to lower the chance of sexual contact with semen, vaginal secretions, or blood.
Never re-use or share needles.
Before you take EVIPLERA
Who must not take it
Together with your doctor, you need to decide whether EVIPLERA is right for you.
Do not take EVIPLERA if you are allergic to:
- tenofovir
- tenofovir DF
- emtricitabine,
- rilpivirine or
- any of the other ingredients of EVIPLERA
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- rash, itching or hives on the skin
- swelling of the face, lips, tongue or other parts of the body
Do not take EVIPLERA if you are already taking any other medicines that contain the same active ingredients. The ingredients of EVIPLERA are listed in the product description section of this leaflet.
Do not take EVIPLERA if you are taking other medicines that contain:
- rilpivirine, unless required for dosage adjustment (e.g. Edurant)
- lamivudine (e.g. Combivir, Zeffix, Kivexa, Trizivir)
- efavirenz (e.g. Stocrin)
- lopinavir/ritonavir (e.g. Kaletra).
- etravirine (e.g. Intelence)
- delavirdine (e.g. Rescriptor)
- nevirapine (e.g. Viramune)
- didanosine (e.g. Videx, Videx EC)
- atazanavir sulfate (e.g. Reyataz)
- tenofovir alafenamide (e.g. Genvoya, Descovy, Odefsey, Biktarvy)
Tell your doctor if you are taking any of the following medicines. Some of these medicines may be obtained without a prescription and some of these may be available under other names. These medicines may alter the amount of EVIPLERA in your blood or EVIPLERA may alter the amount of these medicines in your blood. It is important that you carefully read the package leaflets that are provided with these medicines.
Antiviral Agents (to treat Hepatitis C):
- ledipasvir/sofosbuvir (e.g. Harvoni)
- sofosbuvir/velpatasvir (e.g. Epclusa)
- sofosbuvir/velpatasvir/voxilaprevir (e.g. Vosevi)
Antacids (used to treat stomach ulcers, heartburn or acid reflux):
- aluminium / magnesium hydroxide
- calcium carbonate
Anticonvulsants (to treat epilepsy and prevent seizures):
- carbamazepine (e.g. Tegretol, Teril)
- oxcarbazepine (e.g. Trieptal)
- phenobarbital, phenytoin (e.g. Dilantin)
Antimycobacterials (to treat bacterial infections, including tuberculosis [TB]):
- rifabutin (e.g. Mycobutin)
- rifampicin (e.g. Rifadin/Rimycin)
- rifapentine
Corticosteroids (used in a variety of conditions such as inflammation and allergic reactions):
- dexamethasone when taken by the mouth or injected)
H2-Receptor Antagonists (to treat stomach ulcers or used to relieve heartburn from acid reflux):
- ranitidine (e.g. Zantac)
- famotidine (e.g. Pepcidine, Pepzan)
- cimetidine (e.g. Tagamet, Magicul)
- nizatidine (e.g. Nizac, Tazac)
Macrolide Antibiotics (to treat bacterial infections):
- clarithromycin (e.g. Charihexal, Clarac)
- erythromycin (e.g. E-Mycin, Eryc, EES)
- troleandomycin
Narcotic Analgesic:
- methadone (e.g. Biodone, Physeptone)
Proton Pump Inhibitors (prevent or treat stomach ulcers, heartburn or acid reflux disease):
- omeprazole (e.g. APO-Omeprazole, Meprazol)
- lansoprazole (e.g. Zoton, Lanzopran)
- rabeprazole (e.g. Pariet)
- pantoprazole (e.g. Somac)
- esomeprazole (e.g. Nexium)
Herbal Products: St. Johns wort (Hypericum perforatum):
- St. Johns wort (Hypericum perforatum)
Do not take EVIPLERA to treat your HIV-1 infection if you are also taking adefovir dipivoxil to treat your hepatitis B virus (HBV) infection.
This is not a complete list of medicines that you should tell your doctor about.
Do not take EVIPLERA after the expiry or “use by” date (EXP) printed on the bottle. If you take it after the expiry date has passed, it may not work as well.
Do not take EVIPLERA if the packaging is torn or shows signs of tampering.
Before you start to take it
Tell your doctor if you take an antacid medicine (a medicine to treat heartburn from acid reflux) that contains aluminum, magnesium hydroxide, or calcium carbonate. Take antacids at least 2 hours before or at least 4 hours after EVIPLERA.
Tell your doctor if you take an H2-receptor antagonist (medicines use to treat stomach ulcers, heartburn or acid reflux disease such as cimetidine, famotidine, nizatidine or ranitidine). Take the H2-receptor antagonist at least 12 hours before or at least 4 hours after EVIPLERA. Importantly, proton pump inhibitors (such as omeprazole, lansoprazole, rabeprazole, pantoprazole, esomeprazole) also available for these conditions should not be taken with EVIPLERA.
Tell your doctor if you are allergic to foods, dyes, preservatives or any other medicines.
Tell your doctor if you are pregnant, or planning to become pregnant during your course of medication. We do not know if EVIPLERA can harm your unborn child. You and your doctor will need to decide if EVIPLERA is right for you.
Tell your doctor if you are breastfeeding, or plan to breastfeed during your course of medication. You should not breastfeed if you are HIV-1-positive because of the chance of passing the HIV-1 virus to your baby. Two active substances in this medicine (tenofovir disoproxil fumarate and emtricitabine) have been found in breast milk at low concentrations.
Talk with your doctor about the best way to feed your baby.
Tell your doctor if you have kidney problems or are undergoing kidney dialysis treatment.
Tell your doctor if you have bone problems.
Tell your doctor if you have liver problems, including hepatitis B or C virus infection.
Tell your doctor if you have mental health problems.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines including any that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may affect the levels of EVIPLERA or EVIPLERA may affect the levels of other medicines in the body when they are taken at the same time as EVIPLERA.
Your doctor may change your other medicines or change their doses. Other medicines, including herbal products may affect EVIPLERA.
For this reason, it is very important to let your doctor or pharmacist know what medications, herbal supplements, or vitamins you are taking.
Know the medicines you take. Keep a list of medicines and show it to your doctor and pharmacist when you get a new medicine. Your doctor and your pharmacist can tell you if you can take these medicines with EVIPLERA.
Do not start any new medicines while you are taking EVIPLERA without first talking with your doctor or pharmacist.
How to take EVIPLERA
Take the exact amount of EVIPLERA your doctor has prescribed for you.
Never change the dose on your own.
Do not stop this medicine unless your healthcare provider tells you to stop.
How much to take
The usual dose is one EVIPLERA tablet orally, once daily.
How to take it
Always take EVIPLERA with food. This is important to get the right drug levels in your body.
A nutritional drink alone does not replace food.
If you forget to take EVIPLERA
Do not miss a dose of EVIPLERA.
If you forget to take EVIPLERA within 12 hours of the time you usually take it, take your dose of EVIPLERA with food as soon as possible. Then take your next dose of EVIPLERA at the regularly scheduled time. If you miss a dose of EVIPLERA by more than 12 hours of the time you usually take it, wait and then take the next dose of EVIPLERA at the regularly scheduled time.
Do not take a double dose to make up for a forgotten dose.
When your EVIPLERA supply starts to run low, get more from your doctor or pharmacy. This is very important because the amount of virus in your blood may increase if the medicine is stopped for even a short time. The virus may develop resistance to EVIPLERA and become harder to treat.
Do not change your dose or stop taking EVIPLERA without first talking to your doctor.
If you take too much (overdose)
Immediately telephone your doctor or Poisons Information Centre: 131126 (Australia) and 0800 764 766 (New Zealand) or go to the Accident and Emergency department at your nearest hospital if you think you or anyone else may have taken too many EVIPLERA tablets. Do this even if there are no signs of discomfort or poisoning. This may need urgent medical attention.
While you are taking EVIPLERA
Things you must not do
Do not breast-feed. See “Before you start to take it”
Avoid doing things that can spread HIV infection since EVIPLERA does not stop you from passing the HIV-1 Infection to others:
- Do not share needles or other injection equipment.
- Do not share personal items that can have blood or body fluids on them, like toothbrushes or razor blades.
- Do not have any kind of sex without protection.
Always practice safe sex by using a latex or polyurethane condom or other barrier to reduce the chance of sexual contact with semen, vaginal secretions, or blood.
Things to be careful of
Be careful driving or operating machinery until you know how EVIPLERA affects you. If you are dizzy, have trouble concentrating, or are drowsy, avoid activities that may be dangerous, such as driving or operating machinery.
SIDE EFFECTS
Like all medicines, EVIPLERA can have side effects, although not everybody gets them. Some may be serious and need medical attention.
Check with your doctor as soon as possible if you have any problems while taking EVIPLERA, even if you do not think the problems are connected with the medicine or are not listed in this leaflet.
EVIPLERA may cause the following serious side effects:
Lactic Acidosis
If you have any of the following symptoms after taking your medication, tell your doctor IMMEDIATELY or go to the accident and emergency department at your nearest hospital:
- You feel very weak or tired
- You have unusual (not normal) muscle pain
- You have trouble breathing
- You have stomach pain with nausea and vomiting
- You feel cold, especially in your arms and legs
- You feel dizzy or light headed
- You have a fast or irregular heartbeat
These side effects may be due to a condition called lactic acidosis (build-up of an acid in the blood).
Lactic acidosis can be a medical emergency and may need to be treated in the hospital.
Serious Liver Problems (hepatotoxicity)
If you have any of the following symptoms while taking your medication, tell your doctor IMMEDIATELY or go to the accident and emergency department at your nearest hospital:
- Your skin or the white part of your eyes turns yellow (jaundice)
- Your urine turns dark
- Your bowel movements (stools) turn light in colour
- You don’t feel like eating food for several days or longer
- You feel sick to your stomach (nausea)
- You have lower stomach area (abdominal) pain
These side effects may be due to a condition called hepatotoxicity with liver enlargement (hepatomegaly) andfat deposits in the liver (steatosis) which sometimes occurs in patients taking anti-HIV-1 medicines.
You may be more likely to get lactic acidosis or liver problems if you are female, very overweight (obese), or have been taking similar nucleoside analog-containing medicines, like EVIPLERA, for a long time.
Hepatic Flares
If you have HIV-1 infection and hepatitis B virus (HBV) infection you should not stop your EVIPLERA treatment without first discussing this with your doctor, as some patients have had blood tests or symptoms indicating a worsening of their hepatitis (“hepatic flare”) after stopping individual components (tenofovir DF, and emtricitabine) of EVIPLERA.
You may require medical exams and blood tests for several months after stopping treatment. EVIPLERA is not approved for the treatment of HBV, so you must discuss your HBV therapy with your doctor.
Kidney Problems
If you have had kidney problems in the past or take other medicines that can cause kidney problems, your doctor should do regular blood tests to check your kidneys.
Symptoms that may be related to kidney problems include a high volume of urine, thirst, muscle pain, and muscle weakness.
Changes in Bone Mineral Density (thinning bones)
Laboratory tests show changes in the bones of patients treated with tenofovir DF, a component of EVIPLERA.
Some HIV-1 positive patients treated with tenofovir disoproxil fumarate developed thinning of the bones (osteopaenia) which could lead to fractures.
If you have had bone problems in the past, your doctor may need to do tests to check your bone mineral density or may prescribe medicines to help your bone mineral density.
Additionally, bone pain and softening of the bone (which may contribute to fractures) may occur as a consequence of kidney problems.
Signs and symptoms of inflammation
In some patients with advanced HIV-1 infection (AIDS), signs and symptoms of inflammation from previous infections may occur soon after anti-HIV-1 treatment is started. It is believed that these symptoms are due to an improvement in the body’s immune response, which lets the body fight infections that may have been present with no obvious symptoms.
If you notice any symptoms of infection, please tell your doctor immediately.
Allergy
Some people are allergic to medicines.
If you have any of the following symptoms soon after taking your medicine, DO NOT TAKE ANY MORE EVIPLERA and tell your doctor IMMEDIATELY or go to the accident and emergency department at your nearest hospital.
- shortness of breath
- wheezing or difficulty breathing
- rash, itching or HIV-1es on the skin
- swelling of the face, lips, tongue or other parts of the body
- Mouth sores or blisters on your body
- Inflamed eyes (conjunctivitis)
These are very serious effects. If you have them, you may have a serious allergic reaction. You may need urgent medical attention or hospitalisation.
Pancreatitis
If you have any of the following symptoms after starting your medication, tell your doctor IMMEDIATELY or go to the Accident and Emergency department at your nearest hospital:
- Severe stomach pain or cramps
- Nausea
- Vomiting
These side effects may be due to a condition called pancreatitis which sometimes occurs in patients taking anti-HIV-1 medicines.
Common Side Effects:
Clinical studies have shown the most common side effects of EVIPLERA are:
- depression
- diarrhoea
Other side effects include:
- vomiting
- nausea
- intestinal gas
- dizziness
- allergic reaction
- headache
- sleeping problems (including difficulty to fall asleep or sleepiness)
- abnormal dreams
- stomach pain or discomfort
- indigestion
- rash
- skin discolouration (small spots or freckles)
- pain
- weakness
- decreased appetite and
- fatigue
Marketing experience has shown other side effects reported since emtricitabine and/or tenofovir DF, two components of EVIPLERA, have been marketed include:
- severe allergic reaction (including swelling of the face, lips, tongue, or throat)
- inflammation of the pancreas
- inflammation of the liver
- shortness of breath
- fatty liver
- kidney problems (including decline or failure of kidney function)
- high volume of urine and thirst caused by kidney problems
- muscle pain and muscle weakness
- bone pain, and softening of the bone (which may contribute to fractures) as a consequence of kidney problems have been reported.
- Rash to severe rash and weight gain have also been reported on EVIPLERA.
Ask your doctor or pharmacist if you don’t understand anything in this list.
This is not a complete list of side effects possible with EVIPLERA.
Ask your doctor or pharmacist for a more complete list of side effects of EVIPLERA and all the medicines you will take.
After taking EVIPLERA
Storage
Keep EVIPLERA tablets where children cannot reach them. A locked cupboard at least one-and-a half metres above the ground is a good place to store them.
Keep EVIPLERA tablets in a cool, dry place where it stays below 30 °C.
Do not store EVIPLERA or any other medicine in a bathroom or near a sink.
Do not leave EVIPLERA in the car or on a window sill. Heat and dampness can destroy some medicines.
Keep your EVIPLERA tablets in the bottle with the cap tightly closed until you take them. If you take EVIPLERA tablets out of their pack they may not keep well.
PRODUCT DESCRIPTION
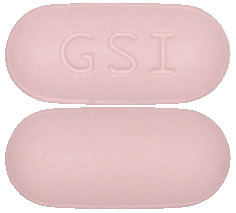
What the tablets look like
EVIPLERA is the brand name of your medicine.
EVIPLERA tablets are capsule-shaped and purplish-pink in colour.
Each tablet is debossed with “GSI” on one side and plain on the other side.
EVIPLERA tablets are supplied in bottles containing 30 tablets.
Ingredients
Each EVIPLERA tablet contains the following active ingredients:
- tenofovir disoproxil fumarate
- emtricitabine
- rilpivirine
Each EVIPLERA tablet also contains the following inactive ingredients:
- pregelatinized starch
- lactose
- cellulose-microcrystalline
- croscarmellose sodium
- magnesium stearate
- povidone
- polysorbate 20
Film-coating:
- macrogol 3350
- hypromellose
- lactose
- glycerol triacetate
- titanium dioxide
- iron oxide red
- indigo carmine aluminium lake
- sunset yellow FCF aluminium lake
SPONSOR
EVIPLERA tablets are supplied in Australia by:
Gilead Sciences Pty Ltd
Level 6, 417 St Kilda Road
Melbourne, Victoria 3004
In New Zealand:
Gilead Sciences (NZ)
c/-Grant Thornton New Zealand Limited
L4, 152 Fanshawe Street,
Auckland 1010
New Zealand
Date of preparation: 15 July 2020
AUST R 176537
BIKTARVY, DESCOVY, EPCLUSA, EVIPLERA, GENVOYA, HARVONI, ODEFSEY, VOSEVI and GSI are trademarks of Gilead Sciences, Inc. or one of its related companies. All other trademarks referenced herein are the property of their respective owners.
Published by MIMS September 2020



 RPV was associated with fewer neurological and psychiatric adverse reactions than efavirenz in patients who received FTC/ tenofovir DF in studies C209 and C215. In addition to the adverse events in studies C209 and C215 (Table 3), the following adverse events were observed in clinical studies of tenofovir DF, FTC and RPV in combination with other antiretroviral agents.
RPV was associated with fewer neurological and psychiatric adverse reactions than efavirenz in patients who received FTC/ tenofovir DF in studies C209 and C215. In addition to the adverse events in studies C209 and C215 (Table 3), the following adverse events were observed in clinical studies of tenofovir DF, FTC and RPV in combination with other antiretroviral agents.
 Virologic outcomes were comparable in males and females in studies C209 and C215.
Virologic outcomes were comparable in males and females in studies C209 and C215. The mean CD4+ cell count increase from baseline to week 24 was 10 cells/mm3 for the Eviplera arm and 22 cells/mm3 for the SBR arm. The difference in median CD4+ cell count change between the Eviplera arm and the SBR arm was not statistically significant at week 24 (p = 0.28).
The mean CD4+ cell count increase from baseline to week 24 was 10 cells/mm3 for the Eviplera arm and 22 cells/mm3 for the SBR arm. The difference in median CD4+ cell count change between the Eviplera arm and the SBR arm was not statistically significant at week 24 (p = 0.28).




