SUMMARY CMI
KIVEXA
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
Important safety information on KIVEXA hypersensitivity reaction is provided in the full CMI. Read before using this medicine.
1. Why am I using KIVEXA?
KIVEXA contains the active ingredients abacavir and lamivudine. KIVEXA is used together with other antiretrovirals to slow down the progression of human immunodeficiency virus (HIV) infection, which can lead to Acquired Immune Deficiency Syndrome (AIDS) and other related illnesses (eg AIDS-related Complex or ARC).
For more information, see Section 1. Why am I using KIVEXA? in the full CMI.
2. What should I know before I use KIVEXA?
Do not use if you have ever had an allergic reaction to abacavir or lamivudine or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use KIVEXA? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with KIVEXA and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use KIVEXA?
- The normal dose for adults and adolescents is one tablet, once a day.
- KIVEXA tablets should be swallowed whole with water.
- KIVEXA tablets do not have to be taken with food.
More instructions can be found in Section 4. How do I use KIVEXA? in the full CMI.
5. What should I know while using KIVEXA?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using KIVEXA? in the full CMI.
6. Are there any side effects?
The most common side effects are feeling sick (nausea), diarrhoea, being sick (vomiting), stomach pain (abdominal pain), headache and rash. Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
PATIENTS TAKING KIVEXA, WHICH CONTAINS ABACAVIR, MAY DEVELOP A HYPERSENSITIVITY REACTION (SERIOUS ALLERGIC REACTION) WHICH CAN BE LIFE-THREATENING IF TREATMENT WITH KIVEXA IS CONTINUED. CONTACT YOUR DOCTOR IMMEDIATELY FOR ADVICE ON WHETHER YOU SHOULD STOP TAKING KIVEXA IF:
1) YOU GET A SKIN RASH OR
2) YOU GET ONE OR MORE SYMPTOMS FROM AT LEAST TWO OF THE FOLLOWING GROUPS:
- FEVER
- SHORTNESS OF BREATH, SORE THROAT OR COUGH
- NAUSEA OR VOMITING OR DIARRHOEA OR ABDOMINAL PAIN
- SEVERE TIREDNESS OR ACHINESS OR GENERALLY ILL FEELING
IF YOU HAVE HAD A HYPERSENSITIVITY (ALLERGIC) REACTION TO KIVEXA TABLETS, NEVER TAKE KIVEXA, OR ANY OTHER MEDICINAL PRODUCT CONTAINING ABACAVIR (TRIUMEQ, TRIZIVIR & ZIAGEN) AGAIN AS YOU MAY DEVELOP A LIFE THREATENING REACTION WHICH CAN BE FATAL.
THERE IS AN ALERT CARD INCLUDED IN THE KIVEXA PACK, TO REMIND YOU AND MEDICAL STAFF ABOUT ABACAVIR HYPERSENSITIVITY. THIS CARD SHOULD BE REMOVED FROM THE PACK AND KEPT WITH YOU AT ALL TIMES. SEE MORE DETAILS UNDER BEFORE YOU TAKE KIVEXA.
FULL CMI
KIVEXA
Active ingredient(s): abacavir (as sulfate) and lamivudine
Consumer Medicine Information (CMI)
This leaflet provides important information about using KIVEXA. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using KIVEXA.
Where to find information in this leaflet:
1. Why am I using KIVEXA?
2. What should I know before I use KIVEXA?
3. What if I am taking other medicines?
4. How do I TAKE KIVEXA?
5. What should I know while using KIVEXA?
6. Are there any side effects?
7. Product details
1. Why am I using KIVEXA?
KIVEXA contains the active ingredients abacavir and lamivudine. KIVEXA is a type of medicine known as an anti-retroviral.
KIVEXA is used to treat Human Immunodeficiency Virus (HIV) infection.
KIVEXA tablets do not cure AIDS or kill the HIV virus, but delays further damage to the immune system by stopping production of new viruses.
You can still pass on HIV when taking this medicine, although the risk is lowered by effective antiretroviral therapy. You will still be able to pass on the HIV virus by sexual activity or by contamination with infected blood. You should still use proper precautions to prevent this. Discuss with your doctor the precautions needed to avoid infecting other people.
KIVEXA tablets are used together with other antiretrovirals to slow down the progression of human immunodeficiency virus (HIV) infection, which can lead to Acquired Immune Deficiency Syndrome (AIDS) and other related illnesses (eg AIDS-related Complex or ARC).
2. What should I know before I use KIVEXA?
Warnings
Do not use KIVEXA if:
- you have ever had an allergic reaction to abacavir, which is also included in medicines called TRIUMEQ, TRIZIVIR and ZIAGEN
- you are allergic to the active ingredient lamivudine, or any of the ingredients listed at the end of this leaflet.
- Always check the ingredients to make sure you can use this medicine.
- if you have a serious liver disease KIVEXA may not be suitable for you
- you have severe kidney disease.
Check with your doctor if you:
- have any other medical conditions
- take any medicines for any other condition
- have allergies to any other medicines, foods, preservatives or dyes
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
Symptoms of infection and inflammation
People with advanced HIV infection (AIDS) have weak immune systems and are more likely to develop serious infections (opportunistic infections). When they start treatment, the immune system becomes stronger, so the body starts to fight infections.
Symptoms of infection and inflammation may develop, caused by either:
- old, hidden infections flaring up again as the body fights them
- the immune system attacking healthy body tissue (autoimmune disorders).
The symptoms of autoimmune disorders may develop many months after you start taking medicine to treat your HIV infection.
Symptoms may include:
- muscle weakness and/or muscle pain
- joint pain or swelling
- weakness beginning in the hands feet and moving towards the trunk of the body
- palpitations or tremor
- hyperactivity (excessive restlessness and movement).
If you get any symptoms of infection or if you notice any of the symptoms above tell your doctor immediately. Don't take other medicines for the infection without your doctors' advice.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Abacavir or lamivudine in KIVEXA tablets may interact with certain other medicines. KIVEXA tablets should not be taken with emtricitabine.
Some medicines may interfere with KIVEXA and affect how it works.
- sorbitol-containing medicines (usually liquids) used regularly
- trimethoprim-sulphamethoxazole (also known as co-trimoxazole), (an antibiotic used to treat Pneumocystis jiroveci pneumonia (often referred to as PCP) or toxoplasmosis).
If you are taking methadone, your doctor may need to adjust your methadone dose, as abacavir increases the rate at which methadone leaves your body. This is unlikely to affect most methadone users.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect KIVEXA.
4. How do I TAKE KIVEXA?
How much to take
- The normal dose for adults and adolescents is one tablet once a day
- Follow the instructions provided and use KIVEXA until your doctor tells you to stop.
When to take KIVEXA
- KIVEXA tablets should be swallowed whole with water.
KIVEXA tablets do not have to be taken with food.
If you forget to take KIVEXA
KIVEXA should be used regularly at the same time each day. If you miss your dose at the usual time, take it as soon as you remember, and then continue as before.
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Do not take a double dose to make up for the dose you missed.
If you take too much KIVEXA
If you think that you have used too much KIVEXA, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
- (by calling 13 11 26), or
- contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using KIVEXA?
Things you should do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking KIVEXA.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine.
It may affect other medicines used during surgery.
If you become pregnant while taking this medicine, tell your doctor immediately.
Keep all of your doctor's appointments so that your progress can be checked.
If you have stopped taking KIVEXA tablets for any reason,
it is important that you contact your doctor before restarting. This is especially so if you think you are having side-effects or have another illness. In some cases your doctor will ask you to restart KIVEXA tablets where medical care can be readily accessed by yourself or others.
You will need regular blood tests
For as long as you're taking KIVEXA, your doctor will arrange regular blood tests to check for side effects.
Stay in regular contact with you doctor
KIVEXA helps to control your condition, but it is not a cure for HIV infection. You need to keep taking it every day to stop your illness from getting worse. Because KIVEXA does not cure HIV infection, you may still develop other infections and illnesses linked to HIV infection.
Keep in contact with your doctor, and don't stop taking KIVEXA without your doctor's advice.
Things you should not do
- Do not give this medicine to anyone else, even if their symptoms seem similar to yours.
- Do not use KIVEXA tablets to treat any other complaints unless your doctor tells you to
- If you have hepatitis B infection, you should not stop
KIVEXA tablets without instructions from your doctor, as you may have a recurrence of your hepatitis. This may occur due to you suddenly stopping lamivudine
Hypersensitivity reaction
- KIVEXA contains abacavir. Abacavir can cause a serious allergic reaction known as a hypersensitivity reaction, which can be life-threatening if treatment with abacavir containing products is not stopped.
- Research has found that people with a gene called HLA-B (type 5701) are more likely to have a hypersensitivity reaction to abacavir. However, even if you do not have this gene type it is still possible for you to get this reaction. If you know you have this gene type, be sure to tell your doctor before you take abacavir.
- The most common symptoms of this reaction include high temperature (fever) and a skin rash. Other most frequently seen symptoms include nausea, vomiting, diarrhoea or abdominal pain; severe tiredness or body aches or generally feeling ill; headache; shortness of breath, sore throat or cough. If you develop any of these symptoms call your doctor IMMEDIATELY WHO WILL ADVISE YOU WHETHER YOU SHOULD STOP TAKING KIVEXA tablets. If your doctor is not available you must urgently seek other medical advice (eg. the Accident and Emergency unit of the nearest hospital) before taking the next dose.
- Other symptoms may include joint or muscle pain, swelling of the neck or itchy skin. Occasionally inflammation of the eye (conjunctivitis), ulcers in the mouth, tingling or numbness of the hands or feet or low blood pressure may occur. The symptoms of this allergic reaction can occur at any time during treatment with KIVEXA tablets. However they usually occur in the first six weeks of treatment, and get worse with continued treatment.
- If you have had this serious reaction to KIVEXA tablets, NEVER take KIVEXA or any other medicinal product containing abacavir (TRIZIVIR, ZIAGEN and TRIUMEQ) again as within hours you may experience a life-threatening lowering of your blood pressure or death.
- Occasionally life threatening hypersensitivity reactions have occurred when KIVEXA tablets was restarted in patients who reported only one of the symptoms on the Alert Card before stopping.
- On very rare occasions, hypersensitivity has been reported when KIVEXA tablets were re-started in patients who had no symptoms of hypersensitivity before stopping.
- If you have stopped taking KIVEXA tablets for any reason it is important that you contact your doctor before restarting. This is especially so if you think you are having side-effects from other medicines or have another illness. Your doctor will check whether any symptoms you had before stopping may be related to this hypersensitivity reaction. If your doctor thinks there is a possibility that they were related, you may be told never to take KIVEXA tablets again. It is important that you follow this advice.
- If you are hypersensitive to KIVEXA tablets you should return all of your unused KIVEXA tablets to your doctor or pharmacist for proper disposal.
Kidney disease
If you have moderate kidney disease you should be monitored as you are at higher risk of some side effects. See additional information under Section 6. Are there any side effects?
Driving or using machines
Be careful before you drive or use any machines or tools until you know how KIVEXA affects you.
No studies on the effects of KIVEXA tablets on the ability to drive and use machines have been performed. However, you should take into account the state of your health and the possible side effects of KIVEXA tablets before considering driving or using machines.
Looking after your medicine
- Keep your tablets in the bottle until it is time to take them.
If you take the tablets out of the bottle they may not keep well. - Keep your tablets in a cool dry place where the temperature stays below 30°C.
- Do not store KIVEXA or any other medicine in the bathroom or near a sink. Do not leave it on a window-sill or in the car. Heat and dampness can destroy some medicines.
- Keep it where young children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
| Very common side effects These may affect at least 1 to 10 in every 100 people: Gastrointestinal:
These may affect less than 1 in every 100 people: From blood tests:
These may affect less than 1 in every 1,000 people General:
These may affect less than 1 in every 10,000 people General:
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
Lactic Acidosis
The symptoms of an allergic (anaphylactic) reaction which may occur soon after starting KIVEXA include wheezing, swelling of the lips/ mouth, difficulty in breathing, hayfever, lumpy rash (hives) or fainting. | Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What KIVEXA contains
| Active ingredient (main ingredient) | 600 mg of abacavir as the sulfate salt and 300 mg of lamivudine. |
| Other ingredients (inactive ingredients) | magnesium stearate microcrystalline cellulose sodium starch glycollate Opadry Orange YS-1-13065-A (containing hypromellose, titanium dioxide CI 77891, macrogol 400, polysorbate 80, sunset yellow FCF CI 15985 aluminium lake) |
Do not take this medicine if you are allergic to any of these ingredients.
What KIVEXA looks like
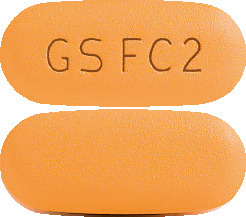
KIVEXA tablets are orange, film-coated, capsule shaped, engraved with GS FC2 on one side. (AUST R 99090).
KIVEXA tablets are supplied in blister packs containing 30 tablets.
Who distributes KIVEXA
KIVEXA is supplied in Australia by:
ViiV Healthcare Pty Ltd
Level 4, 436 Johnston Street
Abbotsford, VIC 3067
Australia
Trademarks are owned by or licenced to the ViiV Healthcare group of companies.
© 2022 ViiV Healthcare group of companies or its licensor.
This leaflet was prepared on 9 May 2022.
Version 11.0
Published by MIMS September 2022


 Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at http://www.tga.gov.au/reporting-problems.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at http://www.tga.gov.au/reporting-problems. The abacavir once daily group was demonstrated to be noninferior when compared to the twice daily group in the overall and baseline viral load subgroups. The incidence of adverse events reported were similar in the two treatment groups.
The abacavir once daily group was demonstrated to be noninferior when compared to the twice daily group in the overall and baseline viral load subgroups. The incidence of adverse events reported were similar in the two treatment groups. Lamivudine has the following structural formula:
Lamivudine has the following structural formula:
