
SUMMARY CMI
LENVIMA®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
▼ This medicine is new or being used differently. Please report side effects. See the full CMI for further details.
1. WHY AM I USING LENVIMA?
LENVIMA contains the active ingredient Lenvatinib (as lenvatinib mesilate). LENVIMA is used to treat patients with thyroid cancer, advanced kidney cancer (advanced renal cell carcinoma), liver cancer or endometrial cancer.
For more information, see Section 1. Why am I using LENVIMA? in the full CMI.
2. WHAT SHOULD I KNOW BEFORE I USE LENVIMA?
Do not use if you have ever had an allergic reaction to LENVIMA or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use LENVIMA? in the full CMI.
3. WHAT IF I AM TAKING OTHER MEDICINES?
Some medicines may interfere with LENVIMA and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. HOW DO I USE LENVIMA?
- The recommended dose of LENVIMA changes depending on which cancer LEVIMA is being used to treat. Your doctor will tell you the correct dose
More instructions can be found in Section 4. How do I use LENVIMA? in the full CMI.
5. WHAT SHOULD I KNOW WHILE USING LENVIMA?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Drinking alcohol |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using LENVIMA? in the full CMI.
6. ARE THERE ANY SIDE EFFECTS?
Tell your doctor straight away if you notice any of the following side effects; you may need urgent medical attention:
- Are feeling numb or weak on one side of your body, severe headache, seizure or fit, confusion, difficulty talking, vision changes or feeling dizzy
- Have chest pain or pressure, pain in your arms, back, neck, jaw, being short of breath, rapid or irregular heart rate, coughing, bluish colour to the lips or fingers, feeling very tired
- Have severe pain in your belly (abdomen)
- Have black, tarry, or bloody stools, or coughing up of blood
- Have yellow skin or yellowing of the whites of the eyes (jaundice) or drowsiness, confusion, poor concentration
- Have diarrhoea, feeling and being sick
- Have pain in the mouth, teeth and/or jaw.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
▼ This medicine is subject to additional monitoring. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. You can report side effects to your doctor, or directly at www.tga.gov.au/reporting-problems.
FULL CMI
LENVIMA®
Active ingredient(s): lenvatinib (as lenvatinib mesilate)
Consumer Medicine Information (CMI)
This leaflet provides important information about using LENVIMA. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using LENVIMA.
Where to find information in this leaflet:
1. Why am I using LENVIMA?
2. What should I know before I use LENVIMA?
3. What if I am taking other medicines?
4. How do I use LENVIMA?
5. What should I know while using LENVIMA?
6. Are there any side effects?
7. Product details
1. Why am I using LENVIMA?
LENVIMA contains the active ingredient Lenvatinib (as Lenvatinib mesilate). LENVIMA is a tyrosine kinase Inhibitor (TKI).
LENVIMA blocks the action of proteins called receptor tyrosine kinases (RTKs), which are involved in the development of new blood vessels that supply oxygen and nutrients to cells and help them to grow. These proteins can be present in high amounts in cancer cells, and by blocking their action LENVIMA may slow the rate at which the cancer cells multiply and the tumour grows and by helping to cut off the blood supply that the cancer needs.
LENVIMA is used to treat thyroid cancer in adults when radioactive iodine treatment has not helped to stop your disease.
It is also used in combination to treat patients with advanced kidney cancer (advanced renal cell carcinoma):
- LENVIMA may be used with the medicine pembrolizumab as your first treatment when your kidney cancer has spread or cannot be removed by surgery.
- LENVIMA may be used with the medicine everolimus after one course of treatment with another anti-cancer medicine.
It is also used to treat liver cancer (hepatocellular carcinoma).
It is also used (along with another medicine called pembrolizumab) to treat a kind of uterine cancer called endometrial carcinoma, if laboratory tests show the cancer is not microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) and other treatments have not helped stop the disease.
2. What should I know before I use LENVIMA?
Before taking LENVIMA, your doctor may carry out some blood tests, for example to check your blood pressure and your liver or kidney function and to see if you have low levels of salt and high levels of thyroid stimulating hormone in your blood. Your doctor will discuss the results of these tests with you and decide whether you can be given LENVIMA. You may need to have additional treatment with other medicines, to take a lower dose of LENVIMA, or to take extra care due to an increased risk of side effects.
Ask your doctor if you have any questions about why this medicine has been prescribed for you.
Warnings
Do not use LENVIMA if:
- you are allergic to Lenvatinib (as Lenvatinib mesilate), or any of the ingredients listed at the end of this leaflet.
- you are breast-feeding.
- Always check the ingredients to make sure you can use this medicine.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Check with your doctor if you:
- Have high blood pressure
- are a woman able to become pregnant
- Have a history of heart problems or stroke
- Are over 75 years of age
- Have had recent surgery or radiotherapy
- Have liver or kidney problems
- have had recent surgery or radiotherapy
- take any medicines for any other condition
- need to have a surgical procedure. Your doctor may consider stopping LENVIMA if you will be undergoing a major surgical procedure as LENVIMA may affect wound healing. LENVIMA may be restarted once adequate wound healing is established.
- have or have had pain in the mouth, teeth and/or jaw, swelling or sores inside the mouth, numbness or a feeling of heaviness in the jaw, or loosening of a tooth. You may be advised to have a dental check-up before starting LENVIMA as bone damage in the jaw (osteonecrosis) has been reported in patients treated with LENVIMA. If you need to undergo an invasive dental treatment or dental surgery, tell your dentist that you are being treated with LENVIMA, particularly when you are also receiving or have received injections of bisphosphonates (used to treat or prevent bone disorders).
- are receiving or have received some medicines used to treat osteoporosis (antiresorptive medicines) or cancer medicines which alter formation of blood vessels (so called angiogenesis inhibitors), as the risk of bone damage in the jaw may be increased.
- belong to an ethnic group other than white or Asian
- weigh less than 60 kg
- have a history of abnormal passageways (known as a fistula) between different organs in the body or from an organ to the skin
- If you have or have had an aneurysm (enlargement and weakening of a blood vessel wall) or a tear in a blood vessel wall.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
LENVIMA is not recommended in pregnancy.
You must use a highly effective method of contraception to avoid becoming pregnant while you are taking LENVIMA. You should continue doing this for one month after stopping treatment.
Do not breast-feed if you are taking LENVIMA. This is because the medicine may pass into breast milk and may seriously harm your breastfed baby.
Children and Teenagers
Do not give this medicine to anyone under the age of 18 years. LENVIMA is not recommended for use in children and teenagers. The effects of LENVIMA in people younger than 18 years old are not known.
Safety and effectiveness in children younger than 18 years have not been established.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with LENVIMA and affect how it works.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect LENVIMA.
4. How do I use LENVIMA?
How much to take / use
Thyroid Cancer
- If you have thyroid cancer, the recommended dose for this medicine is 24 mg once a day (taken as 2 x 10 mg capsules and 1 x 4 mg capsule).
- If you have severe liver or kidney problems your doctor may prescribe a lower dose of 14 mg once a day (taken as 1 x 10 mg capsule and 1 x 4 mg capsule).
Kidney Cancer
- If you have advanced kidney cancer and you have been prescribed Lenvima in combination with pembrolizumab, the recommended daily dose for Lenvima is 20 mg once a day (two 10-mg capsules) in combination with pembrolizumab. Your doctor will also give you an intravenous infusion of pembrolizumab for about 30 minutes every three weeks or every six weeks depending on the dose you are given.
- If you have advanced kidney cancer and you have been prescribed Lenvima in combination with everolimus, the recommended dose for Lenvima is 18 mg once a day (taken as 1 x 10 mg capsule and 2 x 4 mg capsules) in combination with one 5 mg tablet of everolimus once a day.
- If you have severe liver or kidney problems your doctor may prescribe a lower dose of 10 mg once a day (taken as 1 x 10 mg capsule). Your doctor will check to see how much pembrolizumab or everolimus you should receive.
Liver Cancer
- It you have liver cancer. the recommended dose for this medicine is usually 12 mg if your body weight is equal to or more than 60 kg (3 capsules of 4 mg) and 8 mg if your body weight is less than 60 kg ( 2 capsules of 4 mg) once a day.
Endometrial carcinoma
- If you have endometrial carcinoma, the recommended dose for this medicine is 20 mg once a day (taken as 2 x 10 mg capsules). Your doctor will also give you an intravenous infusion of pembrolizumab for about 30 minutes every three weeks or every six weeks depending on the dose you are given.
- If you have severe liver or kidney problems your doctor may prescribe a lower dose of 10 mg once a day (taken as 1 x 10 mg capsule). Your doctor will check to see how much pembrolizumab you should receive.
Your doctor may have prescribed a different dose.
Your doctor may decrease your dose if you have problems with side effects.
Follow the instructions provided and use LENVIMA until your doctor tells you to stop.
Ask your doctor or pharmacist if you are unsure of the correct dose for you. They will tell you exactly how much to take.
Follow the instructions they give you.
If you take the wrong dose, LENVIMA may not work as well and your problem may not improve.
When to take LENVIMA
- LENVIMA should be taken at about the same time each day.
- Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
How to take LENVIMA
Swallow the capsules whole with a full glass of water. If unable to swallow the capsule whole, then place the capsule in a glass of about 25 mL of water or apple juice without breaking or crushing the capsules.
Do not mix more than one medicine in the glass at the same time.
Let the capsules disintegrate for about 10 minutes and then gently stir for at least 3 minutes to dissolve the capsule shells. Then swallow the suspension. After drinking, add the same amount of water or apple juice to the glass and gently swirl it around and swallow the mixture.
The person preparing the suspension should thoroughly wash their hands before and after the suspension is prepared and the dose taken.
You can take LENVIMA with or without food.
Do not chew, crush or split the capsules. To ensure you get the entire dose, the capsules should be swallowed whole without chewing or crushing.
Caregivers should not open capsules to avoid exposure to the contents of the capsule.
If you forget to use LENVIMA
LENVIMA should be used regularly at the same time each day.
If it is 12 hours or more until your next dose, take the missed dose as soon as you remember. Then take the next dose at the normal time. If it is less than 12 hours until your next dose, skip the missed dose. Then take your next dose at the normal time.
Do not take a double dose to make up for the dose that you missed.
This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you use too much LENVIMA
If you think that you have used too much LENVIMA, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using LENVIMA?
Things you should do
Call your doctor straight away if you:
- If you do not feel well while you are taking LENVIMA.
- If you become pregnant while taking LENVIMA. Do not stop treatment without first discussing it with your doctor.
Remind any doctor, dentist or pharmacist you visit that you are using LENVIMA.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking LENVIMA. It may affect other medicines used during surgery.
If you are about to have any blood tests, tell your doctor that you are taking LENVIMA. It may interfere with the results of some tests.
Keep all of your doctor's appointments so that your progress can be checked.
Things you should not do
- Do not stop using this medicine suddenly.
- Do not take LENVIMA to treat any other complaints unless your doctor tells you to.
- Do not give your medicine to anyone else, even if they have the same condition as you.
- Do not stop taking your medicine or lower the dosage without checking with your doctor.
Patients starting LENVIMA should be carefully observed especially when starting treatment and if the dose is increased.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how LENVIMA affects you.
LENVIMA may cause side effects that can affect your ability to drive or use machines.
LENVIMA may make you feel dizzy or sleepy, particularly at the beginning of treatment. If this happens to you, do not drive or use any tools or machines.
Drinking alcohol
Tell your doctor if you drink alcohol.
Avoid alcohol while taking LENVIMA as it may make these effects worse.
Looking after your medicine
- Keep your medicine in the original container.
- If you take it out of its original container it may not keep well
- Keep your medicine in a cool dry place where the temperature stays below 30°C.
Follow the instructions in the carton on how to take care of your medicine properly.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep it where young children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If the medicine has expired or the packing is damaged, return it to your pharmacist for disposal.
When to discard your medicine
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Your doctor may do blood tests to check for side effects.
Other side effects not listed here may occur in some people.
Serious side effects
When you take LENVIMA, you may have some serious side effects. These side effects can sometimes become life-threatening and can lead to death. These side effects may happen anytime during treatment or even after your treatment has ended. You may experience more than one side effect at the same time.
If you have any of the following symptoms, call or see your doctor right away.
| Serious side effects | What to do |
Signs and symptoms of a stroke or mini-stroke, bleeding on your brain or the effect on your brain of a severe increase in blood pressure:
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Less serious side effects
The following side effects have been reported in clinical trials:
| Less serious side effects | What to do |
Hormone related:
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What LENVIMA contains
| Active ingredient (main ingredient) | lenvatinib (as lenvatinib mesilate) |
| Other ingredients (inactive ingredients) | Calcium carbonate Mannitol Cellulose – microcrystalline Hydroxypropylcellulose, Low-substituted hydroxypropylcellulose Talc – purified Hypromellose Titanium dioxide Iron oxide yellow Iron oxide red Shellac Iron oxide black Potassium hydroxide Propylene glycol |
| Potential allergens | none |
Do not take this medicine if you are allergic to any of these ingredients.
What LENVIMA looks like
4 mg hard capsule: A yellowish-red body and yellowish-red cap, approximately 14.3 mm in length, marked in black ink with “Є” on the cap, and “LENV 4 mg” on the body.
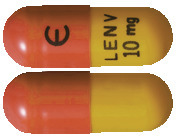
10 mg hard capsule: A yellow body and yellowish-red cap, approximately 14.3 mm in length, marked in black ink with “Є” on the cap, and “LENV 10 mg” on the body.
Australian Register Numbers:
AUST R 233425: LENVIMA lenvatinib 4mg hard capsule blister pack
AUST R 233426: LENVIMA lenvatinib 10mg hard capsule blister pack
Who distributes LENVIMA
Eisai Australia Pty Ltd
Level 2, 437 St Kilda Road
Melbourne, VIC, 3004
[email protected]
This leaflet was prepared in April 2022.
Published by MIMS June 2022




 Table 6 presents laboratory abnormalities occurring in ≥ 20% of patients (All grades) or ≥ 3% (Grades 3-4) of patients with Lenvima in combination with pembrolizumab.
Table 6 presents laboratory abnormalities occurring in ≥ 20% of patients (All grades) or ≥ 3% (Grades 3-4) of patients with Lenvima in combination with pembrolizumab.
 In Table 8, Grade 3-4 laboratory abnormalities occurring in ≥ 3% of patients in the Lenvima with everolimus arm are presented.
In Table 8, Grade 3-4 laboratory abnormalities occurring in ≥ 3% of patients in the Lenvima with everolimus arm are presented.
 Table 10, Grade 3-4 laboratory abnormalities occurring in ≥ 2% of patients in the Lenvima arm in REFLECT (HCC) are presented.
Table 10, Grade 3-4 laboratory abnormalities occurring in ≥ 2% of patients in the Lenvima arm in REFLECT (HCC) are presented.


















