


What is in this leaflet
This leaflet answers some common questions about PRILACE.
It does not contain all of the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have benefits and risks. Your doctor has weighed the risks of you taking PRILACE against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, talk to your doctor or pharmacist.
Keep this leaflet with your medicine. You may need to read it again.
What PRILACE is used for
PRILACE contains ramipril, which belongs to a group of medicines called angiotensin-converting enzyme (ACE) inhibitors.
PRILACE is used to treat:
- high blood pressure (hypertension)
- some heart conditions such as heart failure after a heart attack
- kidney problems in some patients.
PRILACE is also used to reduce the risk of cardiovascular problems and complications in patients aged 55 years or more with heart or blood vessel disease, or diabetes.
Hypertension
PRILACE is used to lower high blood pressure (hypertension). Everyone has blood pressure. This pressure helps circulating your blood around your body. Your blood pressure is different at different times of the day and can be affected by how busy or worried you are. You would have hypertension when your blood pressure stays higher than is needed, even when you are calm and relaxed.
There are usually no symptoms of hypertension. The only way of knowing you have hypertension is to have your blood pressure checked on a regular basis. If hypertension is not treated, it can lead to serious health problems, including stroke, heart disease and kidney failure.
Heart failure after a heart attack
PRILACE may be used after a heart attack. A heart attack occurs when one of the major blood vessels supplying blood to your heart becomes blocked. This means that your heart muscle cannot receive the oxygen it needs and becomes damaged. This may lead to further problems, such as heart failure, irregular heart rhythms and blood clots.
Heart failure means that the heart muscle is weak and cannot pump blood strongly enough to supply all the blood needed throughout the body. Heart failure is not the same as heart attack and does not mean that the heart stops. Heart failure may start off with no symptoms, but as the condition progresses, patients may feel short of breath or may get tired easily after light physical activity such as walking. Some patients may wake up at night with difficulty breathing. Fluid may collect in different parts of the body, often first noticed as swollen ankles and feet.
Kidney problems
PRILACE may be used to treat some kidney problems. Some conditions, such as diabetes and hypertension, can lead to kidney problems. These problems develop slowly over several years. Good control of your blood sugar and blood pressure are important in keeping your kidneys healthy, but may not always prevent kidney damage from occurring.
Prevention of cardiovascular problems and complications PRILACE may be used to reduce the risk of some cardiovascular problems and complications in patients aged 55 or more who have:
- coronary artery disease (heart disease caused by poor blood flow in the blood vessels of the heart)
- peripheral vascular disease (poor circulation in the hands or feet)
- stroke.
PRILACE may also be used to reduce the risk of cardiovascular problems and complications in diabetic patients aged 55 years or more who have:
- high blood pressure
- high cholesterol levels
- kidney problems
- previous blood vessel disease
- and are current smokers.
How PRILACE works
PRILACE works by widening the blood vessels, which reduces the pressure in the vessels, making it easier for your heart to pump blood around your body. This helps increasing the supply of oxygen to your heart, so that when you place extra demands on your heart, such as during exercise, your heart may cope better and you may not get short of breath as easily.
By increasing the supply of oxygen to your heart, your heart does not have to work as hard and it is under less stress, which may reduce the risk of further damage occurring to it following a heart attack.
PRILACE also improves blood flow to the small blood vessels found in the kidneys, in which helps the kidneys to work more efficiently. This in turn can help to slow down the progression of kidney damage that might result from having diabetes or high blood pressure.
Ask your doctor if you have any questions about why PRILACE has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is not addictive.
It is available only with a doctor's prescription.
Before you take it
When you must not take it
Do not take PRILACE if you are allergic to ramipril, or any of the ingredients listed at the end of this leaflet. Symptoms of an allergic reaction may include skin rash, itching, difficulty breathing, swelling of the face, lips or tongues, abdominal pain, muscle or joint pain.
Do not take PRILACE if you have previously taken any other ACE inhibitor medicines that caused your face, lips, tongue or throat to swell, or made it hard for you to breathe. If you have had an allergic reaction to an ACE inhibitor before, you may be allergic to PRILACE.
Do not take PRILACE if you or your family have a history of swelling of the face, lips, tongue, throat, intestines, hands or feet for no apparent reason.
Do not take PRILACE if you have a kidney problem known as renal artery stenosis.
Do not take PRILACE if you have problems or conditions affecting the flow of blood in and out of your heart such as aortic or valvular stenosis.
Do not take PRILACE if you are pregnant or intend to become pregnant. PRILACE may affect your developing baby if you take it during pregnancy.
Do not take PRILACE if you are breastfeeding. PRILACE may pass into breast milk and affect your breast-fed baby.
Do not take PRILACE if you undergo dialysis using certain high-flux membranes.
Do not take PRILACE if you have low blood pressure.
Do not take PRILACE if the expiry date (Exp.) printed on the pack has passed.
Do not take PRILACE if the packaging is torn or shows signs of tampering.
If you are not sure whether you should start taking PRILACE, talk to your doctor.
Before you start to take it
Tell your doctor if you are allergic to any other medicines, foods, dyes or preservatives.
Tell your doctor if you have or have had any other medical conditions including:
- kidney problems, or having dialysis (note that your doctor may give you PRILACE because of your kidney problems)
- liver problems
- heart problems (note that your doctor may give you PRILACE because of your heart problems)
- diabetes (note that your doctor may give you PRILACE because of your diabetic problem)
- low blood pressure, which you may notice as dizziness or light-headedness
- low white blood cell counts
- high level of potassium in your blood
- systemic lupus erythematosus, scleroderma or other auto-immune conditions.
Tell you doctor if you have a family history of swelling of the face, lips, tongue, throat, intestines, hands or feet.
Tell your doctor if you:
- follow a very low or very high salt diet
- are dehydrated, or have had a recent bout of vomiting or diarrhoea
- are about to have surgery or a general anaesthetic
- plan to become pregnant or breastfeed.
If you have not told your doctor about any of the above, tell them before you start taking PRILACE.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including those you buy without a prescription from a pharmacy, supermarket or health food shop.
Some medicines may be affected by PRILACE, or may affect how well it works. These include:
- other medicines used to treat high blood pressure
- diuretics, also known as fluid or water tablets
- anti-inflammatory drugs (e.g. non-steroidal anti-inflammatory drugs, COX-2 inhibitors) used to relieve pain, inflammation, swelling and other symptoms including arthritis
- potassium supplements or potassium-containing salt substitutes
- lithium, a medicine used to treat mood swings and some types of depression
- insulin and tablets used to treat diabetes
- heparin
- general anaesthetic
- medicines that may affect blood cells, such as allopurinol, procainamide, corticosteroids, immunosuppressants, or medicines used to treat cancer
- medicines for appetite control, asthma (high doses of corticosteroids), colds, coughs or sinus problems, particularly when PRILACE is used to control high blood pressure.
If you are not sure whether you are taking any of these medicines, check with your doctor or pharmacist. Your doctor can tell you what to do if you are taking any of these medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking PRILACE.
How to take it
How much to take
Your doctor will decide how many tablets or capsules you will need to take each day. This depends on the condition being treated and whether or not you are taking any other medicines. Some patients may need a lower starting dose.
The usual dose of PRILACE is:
- for high blood pressure, 2.5 mg to 10 mg per day
- for heart failure, 5 mg to 10 mg per day
- for kidney problems, 1.25 mg to 5 mg per day
- for cardiovascular risk, 2.5 mg to 10 mg per day.
Depending on your response, your doctor may adjust the dose.
If two tablets are prescribed, your doctor may want you to take them either together or at different times. This will depend on the condition being treated and how you respond to PRILACE.
Follow all directions given to you by your doctor and pharmacist carefully. They may differ from the information contained in this leaflet.
How to take it
PRILACE should be swallowed with plenty of fluid. It can be taken before or after meals.
PRILACE capsules should be swallowed whole.
When to take it
Take PRILACE at about the same time of each day. Taking your tablets or capsules at about the same time of each day will have the best effect. It will also help you to remember when to take it.
How long to take it
Continue taking PRILACE for as long as your doctor tells you. PRILACE helps control your condition but does not cure it. Thus, you must take it every day.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take the missed dose as soon as you remember and then go back to taking your tablets or capsules as you would normally.
Do not take a double dose to make up for the dose you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much PRILACE.
Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
If you take too much PRILACE, you may feel dizzy, light-headed, weak or you may faint. You may also experience slow heartbeat.
While you are taking it
Things you must do
Tell all the doctors, dentists and pharmacists who are treating you that you are taking PRILACE.
Before starting any new medicine, tell your doctor or pharmacist that you are taking PRILACE.
Tell your doctor if you feel light-headed or dizzy after taking your first dose of PRILACE or when your dose is increased.
Tell your doctor immediately if you become pregnant or intend to become pregnant while taking PRILACE.
Make sure you drink enough water during exercise and in hot weather when you are taking PRILACE, especially if you sweat a lot. If you do not drink enough water while taking PRILACE on hot days or during exercising, you may feel dizzy, light-headed or sick. This is because your blood pressure has dropped too much.
If you continue to feel unwell, tell your doctor.
Tell your doctor if you have excess vomiting or diarrhoea while taking PRILACE. You may lose too much water and salt, and your blood pressure may drop too much.
If you plan to have surgery that needs a general anaesthetic, tell your doctor or dentist that you are taking PRILACE. Your blood pressure may drop suddenly during anaesthesia.
If you are about to have any blood tests, tell your doctor that you are taking PRILACE. PRILACE may interfere with the results of some tests.
Go to your doctor regularly for a check-up. Have your blood pressure checked regularly either by your doctor or as instructed. This is to make sure PRILACE is working for your conditions. Also, your doctor may occasionally do a blood test to check your potassium level and see how your kidneys are working.
Things you must not do
Do not stop taking PRILACE or lower or increase the dosage, without checking with your doctor.
Do not use PRILACE to treat any other conditions unless your doctor tells you to.
Do not give PRILACE to anyone else, even if they have the same condition as you.
Things to be careful of
If you feel light-headed or dizzy when getting out of bed or standing up, get up slowly. Standing up slowly, especially when you get up from bed or chairs, will help your body get used to the change in position and blood pressure. If this problem continues or gets worse, talk to your doctor.
Be careful driving or operating machinery until you know how PRILACE affects you. This medicine may cause dizziness, light-headedness, tiredness or drowsiness in some people. Make sure you know how you react to PRILACE before you drive, operate machinery or do anything that could be dangerous if you are dizzy or light-headed. If these occur, do not drive or operate machinery.
If you drink alcohol, dizziness or light-headedness may be worse.
Things that may help your blood pressure or heart failure
Some self-help measures suggested below may help your condition. Talk to your doctor or pharmacist about these measures and for more information.
- Alcohol - your doctor may advise you to limit your alcohol intake.
- Smoking - your doctor may advise you to stop smoking or at least cut down.
- Exercise - regular exercise helps reduce blood pressure and helps get the heart fitter, but it is important not to overdo it. Walking is a good exercise, but try to find a route that is reasonably flat. Before starting any exercise, ask your doctor for a program that best suits you.
- Weight - your doctor may suggest you to lose some weight, so to help lower your blood pressure and lessen the amount of work your heart has to do. Some people may need a dietician’s help to lose weight.
- Diet - eat a healthy low-fat diet that includes plenty of fresh vegetables, fruit, bread, cereals and fish. Also, eat less fat and sugar.
- Salt - your doctor may advise you to watch the amount of salt in your diet. To reduce salt intake, you can avoid or minimise using salt in cooking or at the table.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking PRILACE. Like all medicines, PRILACE may have unwanted side effects in some people. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice any of the following and they worry you:
- feeling dizzy, light-headed, faint
- dry cough
- headache
- nausea (feeling sick), vomiting
- stomach pain
- diarrhoea
- unusual tiredness, weakness
- ringing or buzzing in the ears
- forgetfulness
- confusion.
These side effects are usually mild.
Tell your doctor as soon as possible if you notice any of the following:
- disturbed vision
- symptoms of sunburn (such as redness, itching, swelling, blistering), which may occur more quickly than normal
- itchy or raised skin rash, hives or nettlerash
- yellowing of the skin and/or eyes
- fast or irregular heartbeat
- shortness of breath or tightness in the chest
- severe upper stomach pain, often with nausea and vomiting
- frequent infections such as fever, severe chills, sore throat or mouth ulcers
- passing little or no urine, or more urine than is normal for you
- signs of anaemia such as tiredness, being short of breath and looking pale
- numbness, tingling and colour change (white, blue then red) in the fingers or toes when exposed to the cold
- bleeding or bruising more easily than normal.
These side effects are rare but serious. You may need medical attention.
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you notice any of the following:
- fainting within a few hours of taking a dose
- chest pain
- severe dizziness and confusion with visual disturbances and speech problems
- swelling of the face, lips, mouth, tongue or throat, which may cause difficulty in swallowing or breathing
- pink or red itchy spots on the skin, which may blister and progress to form raised, red, pale-centred marks
- severe blisters and bleeding in the lips, eyes, mouth, nose and genitals.
These are very serious side effects and require immediate attention.
Tell your doctor if you notice anything else that is making you feel unwell. Other side effects not listed above may also occur in some people.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
After taking it
Storage
Keep PRILACE where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Keep PRILACE in the pack until it is time to take them. If you take the medicine out of the pack, it may not keep as well.
Keep PRILACE in a cool dry place, protected from light, where the temperature stays below 25°C.
Do not store PRILACE or any other medicines in the bathroom or near a sink. Do not leave PRILACE in the car or on window sills. Heat and dampness can destroy some medicines.
Disposal
If your doctor tells you to stop taking PRILACE, or the tablets or capsules have passed their expiry date, ask your pharmacist what to do with any that are left over.
Product description
What it looks like
PRILACE is available in four strengths:
- 1.25 mg tablet: white to off-white, oval-shaped tablet marked “RP 1” on one side
- 2.5 mg tablet: white to off-white, oval-shaped tablet marked “RP 2” on one side and scoreline on other side
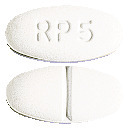
- 5 mg tablet: white to off-white, oval-shaped tablet marked “RP 5” on one side and scoreline on other side
- 10 mg capsule: white and blue opaque capsules, with “RP 10” printed in black.
PRILACE is available in blister packs of 30 tablets or capsules.
Ingredients
PRILACE tablets contain either 1.25 mg, 2.5 mg or 5 mg of the active ingredient ramipril. The tablets also contain:
- sodium bicarbonate
- calcium sulfate
- pregelatinised maize starch
- sodium stearylfumarate.
PRILACE capsules contain 10 mg of the active ingredient ramipril. The capsules also contain pregelatinised maize starch. The capsule shells are size#4 Hard Gelatin Capsules White Op/ Blue Op with black printing ink.
PRILACE tablets and capsules do not contain gluten, sucrose, tartrazine or any azo dyes.
PRILACE capsules may contain traces of sulfites.
Sponsor
Arrotex Pharmaceuticals Pty Ltd
15-17 Chapel Street
Cremorne VIC 3121
www.arrotex.com.au
Australian registration numbers:
PRILACE 1.25 mg: AUST R 121854
PRILACE 2.5 mg: AUST R 121855
PRILACE 5 mg: AUST R 121856
PRILACE 10 mg: AUST R 133085.
This leaflet was revised in December 2022.
Published by MIMS February 2023
 The improvement in these key endpoints was observed to increase with time, to be maintained long term and to apply to both hypertensive and nonhypertensive patients. A delay of approximately three months was seen prior to detection of the beneficial effects of ramipril, suggesting the value of early treatment.
The improvement in these key endpoints was observed to increase with time, to be maintained long term and to apply to both hypertensive and nonhypertensive patients. A delay of approximately three months was seen prior to detection of the beneficial effects of ramipril, suggesting the value of early treatment.
 The results of the HOPE study in terms of the prespecified secondary endpoints for the whole population (intention to treat) are presented in Table 4.
The results of the HOPE study in terms of the prespecified secondary endpoints for the whole population (intention to treat) are presented in Table 4. The results of the HOPE study in terms of the prespecified secondary endpoints for those patients with diabetes are presented in Table 5.
The results of the HOPE study in terms of the prespecified secondary endpoints for those patients with diabetes are presented in Table 5.
 Its empiric formula is C23H32N2O5, and its molecular weight is 416.5.
Its empiric formula is C23H32N2O5, and its molecular weight is 416.5.