What is in this leaflet
This leaflet answers some common questions about Roferon-A pre-filled syringes.
It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using Roferon-A against the benefits they expect it will have for you.
If you have any concerns about using this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine.
You may need to read it again.
What Roferon-A is used for
Roferon-A contains the active ingredient interferon alfa-2a.
Roferon-A belongs to a group of medicines called interferons.
Roferon-A is a similar form of interferon to one of the interferons made naturally by your own immune system as a defence against viruses and tumours. Roferon-A works by slowing the growth of certain tumour cells and viruses and also by boosting the effects of your own immune system to fight these.
Roferon-A is used in the treatment of viral infections of the liver (chronic active hepatitis B and chronic hepatitis C).
Roferon-A treatment will not prevent a hepatitis B or C infected person from giving another person hepatitis B or C.
Roferon-A may be used in combination with ribavirin for the treatment of chronic hepatitis C in patients who have relapsed to previous interferon monotherapy treatment.
Roferon-A is also used in the treatment of various types of cancer such as, kidney cancer (renal cell carcinoma), cancer of the lymph glands (cutaneous T-cell lymphoma), blood cancer (hairy cell leukaemia, chronic myelogenous leukaemia (CML) and other bone marrow conditions causing platelet disorders), AIDS-related cancer (Kaposi's sarcoma), and low-grade non-Hodgkin's lymphoma.
Roferon-A may be used in combination with AVASTIN for the treatment of kidney cancer.
Your doctor, however, may have prescribed Roferon-A for another reason.
Ask your doctor if you have any questions why Roferon-A has been prescribed for you.
Roferon-A is not addictive.
This medicine is available only with a doctor's prescription.
Before you use Roferon-A
Do not use Roferon-A if:
- you have had an allergic reaction to interferon alfa or any ingredients listed at the end of this leaflet
Some symptoms of an allergic reaction include:
- shortness of breath
- wheezing or breathing difficulties
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
- you have advanced liver disease or suffer from autoimmune hepatitis
- the patient is a new born baby - one of the inactive ingredients of Roferon-A, benzyl alcohol, can be harmful to newborns
- the package is torn or shows signs of tampering
- the expiry date (EXP) printed on the pack has passed
If you use this medicine after the expiry date has passed it may have no effect at all, or worse, an unexpected effect.
Before starting Roferon-A, your doctor will discuss with you the possible benefits and possible side effects of treatment, to decide if Roferon-A is right for you. While taking Roferon-A, you will need to see your doctor regularly for medical examinations and blood tests to make sure your treatment is working and to check for side effects.
Use in children
Do not give Roferon-A to children under 18 years of age.
Safety and effectiveness in children have not been established.
This product contains benzyl alcohol and should not be given to babies from birth up to the age of 3 years.
Before you start to use Roferon-A
Your doctor must know about all the following before you start to use Roferon-A.
Tell your doctor if:
- you are pregnant or plan to become pregnant
It is not known whether Roferon-A is harmful to an unborn baby when used by a pregnant woman. If there is a need to use Roferon-A when you are pregnant your doctor will discuss the risks and benefits to you and the unborn baby. - you are breast-feeding or plan to breast-feed
Because interferon alfa-2a occurs naturally in the body, it has not been possible to determine whether Roferon-A passes into the breast milk following injection. Your doctor will discuss the risks and benefits of using Roferon-A if you are breast-feeding. - you have ever had any of the following medical conditions or problems, tell your doctor before you start taking Roferon-A:
- history of, or current severe mental illness (such as depression or anxiety)
- history of injecting drug use or excessive alcohol use
- history of heart disease or previous heart attack
- autoimmune disease (where the body's immune system attacks the body's own cells), such as psoriasis (a skin disease)
- kidney problems
- blood disorders
- diabetes (high blood sugar)
- problems with the thyroid gland
- liver problems, other than hepatitis C
- vision problems
- an organ transplant (e.g. kidney or bone marrow) or an organ transplant is planned for you in the immediate future.
- you are allergic to any other medicines, foods, dyes or preservatives.
Taking other medicines
Tell your doctor if you are taking any other medicines, including any that you have bought from a pharmacy, supermarket or health food shop.
Some medicines may interfere with Roferon-A or may affect how well it works. You may need to use different amounts of your medicine, or you may need to take different medicines. Your doctor will advise you.
You must tell your doctor if you are taking:
- theophylline, a medicine used in the treatment of asthma, bronchitis, emphysema and related conditions.
- sho-saiko-to, a Chinese herbal medicine, also known as Xiao-Chai-Hu-Tang
Your doctor or pharmacist has more information on medicines to be careful with or avoid while using Roferon-A.
If you have not told your doctor about any of the above, tell them before you start using Roferon-A.
How to use Roferon-A
Follow all directions given to you by your doctor or pharmacist carefully.
Those directions may differ from the information contained in this leaflet.
How much to inject
The dosage of Roferon-A is expressed in millions of international units (MIU). If this unitage seems unusually high, it may be helpful to convert it into units that you are more familiar with. For example, 200 MIU adds up to only 1 milligram of Roferon-A.
The dose of Roferon-A will be determined by your doctor based on your condition. As a general rule, your dosage should not exceed 36 MIU per day.
Your dose may be adjusted by your doctor during therapy according to your response.
Your doctor will keep track of your response to Roferon-A by asking questions and performing occasional laboratory tests.
Do not exceed or change the dose recommended by your doctor.
The dosage is generally adjusted depending on your response to treatment.
Directions for administration
Roferon-A is administered by a subcutaneous injection (under the skin).
If you are receiving other treatments for your disease (e.g. chemotherapy, AVASTIN), your Roferon-A injection may be given by a doctor or nurse whilst you are in hospital receiving your other treatment.
It may be appropriate and more convenient for you to receive your injections at home. Your doctor will discuss this with you. If this is the case, you or a family member will be instructed on how to give the injection properly. This is a simple procedure and many patients prefer to administer their treatment at home. Administration of the subcutaneous injection is described further on in this leaflet.
How long to use Roferon-A
Your treatment period could range from a few weeks up to 12 months depending on your illness.
If your injections are administered at a hospital with another treatment (e.g. chemotherapy, AVASTIN), your Roferon-A treatment might continue for several months after your other treatment has stopped. At this time, you may start to self-administer Roferon-A as described below. Your doctor will advise you.
Continue using Roferon-A until your doctor tells you to stop.
Your doctor will determine when your treatment should be stopped.
Directions for self-administration
If you wish to administer Roferon-A yourself at home, please follow the instructions below.
Subcutaneous (sc) Injection
You should read these directions from beginning to end before starting so that each step of the procedure becomes familiar. These instructions must be carefully followed. Consult your doctor if you require further instructions.
Remember that cleanliness is vital.
How to inject
- Before subcutaneous injection
- Check the expiry date. Do not use Roferon-A after the expiry date shown on the pre-filled syringe label.
- Check the dose that you have been prescribed.
- Check the liquid has no discolouration, cloudiness or particles. The liquid should look clear and colourless to slightly yellow.
- Let the syringe stand for 30 minutes at room temperature.
- Wash your hands thoroughly.
- Place everything you need within easy reach: the pre-filled syringe, the needle for subcutaneous injection, an alcohol swab and some cotton wool and a container for disposal of the needle and syringe.
- How to prepare the syringe and needle for injection
A. Take the sealed needle in both hands and snap the grey cap backwards. Remove the cap. DO NOT remove the plastic needle shield.
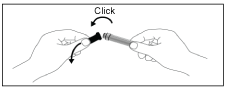
B. Remove the rubber cap from the syringe. Do not touch the tip of the syringe.
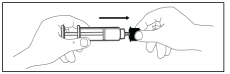
C. Attach the needle with the plastic shield firmly to the syringe.
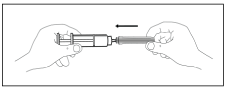
D. Remove the plastic cover from the needle while holding the syringe. Avoid pushing the plunger.
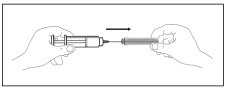
E. To remove air bubbles from the syringe, hold the syringe with the needle pointing up. Tap the syringe gently to bring the bubbles to the top. Push up the plunger slowly to the correct dose. Replace the needle guard and place the syringe in a horizontal position on a clean flat surface.
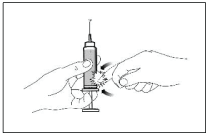
The syringe is now ready for use.
- Performing subcutaneous injection
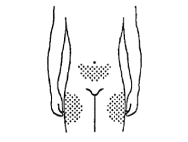
F. Select an injection site in the abdomen or thigh area (except your navel or waistline, see diagram). Change your injection site each time.
G. Remove an alcohol swab from one packet and clean and disinfect the site by wiping with the swab.
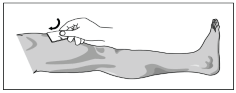
H. Remove the alcohol swab from the site. Allow the injection site to dry for 10 seconds.
I. Remove the needle guard
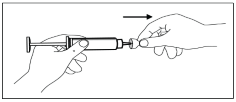
J. With one hand, pinch a fold of loose skin. With your other hand, hold the syringe as you would a pencil.
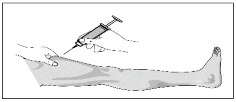
K. Gently insert the needle into the pinched skin at an angle of 45° to 90° (bevelled edge facing upwards). Half of the needle should still be visible.
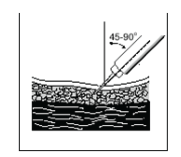
L. Inject the solution by gently pushing the plunger all the way down, while keeping the skin grasped.
M. After injecting, remove the needle at the same angle so it does not hurt you, and release the skin.
Press gently over the injected area with some cotton wool. Don't worry if you see slight bleeding, it should not bleed a lot. If it does bleed, keep pressing on the area for a while. You may need to put a Band-Aid on. Check the site later for any signs of soreness, and if this occurs tell your doctor or nurse at the next opportunity.
Remember:
Most people can learn to give themselves a subcutaneous injection, but if you experience difficulty, please do not be afraid to ask for help and advice from your doctor, nurse or pharmacist.
- How to dispose of used needles and syringes
The needle and syringe are to be used once only. Dispose of the needle and syringe immediately after injection into a sturdy glass or hard plastic container away from children. Do not replace the needle cover.
Never put used needles and syringes into your normal household waste bin. If you are not sure how to dispose of the needles and syringes, consult your doctor, nurse or pharmacist on how to properly dispose the syringes and needles.
When to inject Roferon-A
Your doctor will tell you how often to use this medicine. As a general rule, a maximum of one injection per day should be given.
If you are injecting this medicine yourself, use it at bedtime as Roferon-A may cause unusual tiredness or flu-like symptoms.
If you forget to use Roferon-A
If it is almost time for your injection, skip the injection you missed and use your next injection when you are meant to.
Otherwise, use it as soon as you remember, and then go back to using it as you would normally.
Do not use a double injection to make up for the injection that you missed.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering when to use your medicine, ask your pharmacist for some hints.
If you use too much Roferon-A (overdose)
Immediately telephone your doctor or Poisons Information Centre (telephone 13 11 26) for advice or go to Accident and Emergency at your nearest hospital if you think that you or anyone else may have used too much Roferon-A, even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Keep telephone numbers for these places handy.
While you are using Roferon-A
Things you must do
Use Roferon-A exactly as your doctor has prescribed.
Tell all doctors, dentists and pharmacists who are treating you that you are using Roferon-A.
Do not take any other medicines whether they require a prescription or not without first telling your doctor or consulting with a pharmacist.
Tell your doctor if you become pregnant while using Roferon-A.
Tell your doctor if, for any reason, you have not used Roferon-A exactly as prescribed.
Otherwise, your doctor may think that it was not effective and change your treatment unnecessarily.
Be sure to keep all of your appointments with your doctor so that your progress can be checked.
Things you must not do
Do not stop using Roferon-A or change the dose without first checking with your doctor.
Do not let yourself run out of medicine over the weekend or on holidays.
Do not give Roferon-A to anyone else even if they have the same condition as you.
Do not use Roferon-A to treat other complaints unless your doctor says to.
Do not switch to any other brands of interferon without consulting your doctor because a change in dosage may result.
Do not take any other medicines whether they require a prescription or not without first telling your doctor or consulting a pharmacist.
Things to be careful of
Be careful driving or operating machinery until you know how Roferon-A affects you.
Your doctor may want you to drink extra fluids, especially when you begin Roferon-A treatment.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are using Roferon-A.
Roferon-A helps most people but it may have unwanted side effects in a few people.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you notice any of the following:
- signs of anaemia, such as tiredness, shortness of breath when exercising, headaches, dizziness and looking pale
- signs of mental health problems such as, irritability (getting easily upset), depression (feeling low, bad about yourself, hopeless, very depressed), suicidal, anxiety, aggressive behaviour
- mania (episodes of overactivity, elation or irritability, excitement, over-activity and uninhibited behaviour)
- chest pain or irregular heartbeat
- confusion, depression, trouble sleeping, thinking or concentrating
- numbness or tingling sensation
- coughing and/or breathlessness
- trouble breathing, pneumonia
- fluid build up in body tissues (swelling)
- visual disturbances such as blurred or loss of vision
- convulsions (epileptic fits)
- frequent infections such as fever, severe chills, sore throat or mouth ulcers; unusual bleeding, bruising more easily than usual or nosebleeds
- symptoms of bowel inflammation including diarrhoea (usually with blood or mucus), stomach pain, fever
These are serious side effects. You may need urgent medical attention. Serious side effects are rare.
Tell your doctor if you notice any of the following and they worry you:
- flu-like symptoms such as fever, tiredness, chills, muscle or joint pain and headache (these symptoms can usually be relieved by paracetamol)
- loss of appetite, taste change or dry mouth
- tiredness or sleepiness (may be less troublesome if you have your injection in the evening or before going to sleep)
- dizziness
- nausea (feeling as if you are about to vomit), vomiting or diarrhoea
- heart burn
- slight hair loss (usually temporary and reversible after finishing treatment)
- mild or moderate abdominal (gut) pain
- rash
- dry or itchy skin
- weight loss
- night sweats
- temporary impotence
- reappearance of cold sores if previously infected with herpes virus
This is not a complete list of all possible side effects. Your doctor or pharmacist has a more complete list. Others may occur in some people and there may be some side effects not yet known.
Tell your doctor if you notice anything else that is making you feel unwell, even if it is not on this list.
Ask your doctor or pharmacist if you don't understand anything in this list.
Do not be alarmed by this list of possible side effects.
You may not experience any of them.
After using Roferon-A
Storage
Always keep this medicine closed in its original pack.
If you take the pre-filled syringes out of the pack they may not keep well.
Store Roferon-A pre-filled syringes in the fridge at 2°C to 8°C. Do not freeze.
Protect Roferon-A pre-filled syringes from light.
Do not leave it in the car or on window sills.
Heat and dampness can destroy some medicines.
Keep Roferon-A where young children cannot reach it.
The top shelf of the refrigerator is a good place to store this medicine.
Roferon-A pre-filled syringes are for single use only.
The pre-filled syringe should be used once only and any remaining contents should be discarded with the needle.
Disposal
If your doctor tells you to stop using Roferon-A, or the pre-filled syringe has passed its expiry date, ask your pharmacist what to do with any pre-filled syringes that are left over.
If you use Roferon-A at home, you must throw away the syringes and needles in a sturdy glass or hard plastic container that will not let the needles stick through it.
This will help protect you and other people from accidental needle stick injuries. Being struck by a needle can pass diseases onto other people.
Product description
Availability
Roferon-A pre-filled syringes come in packs of one.
What Roferon-A pre-filled syringes look like
Roferon-A solution for injection is contained in a disposable glass syringe. The solution is clear and colourless to slightly yellowish.
A stainless steel needle is also supplied with the syringe to allow for subcutaneous injection.
Ingredients
Active ingredient
- interferon alfa-2a (rbe)
Roferon-A pre-filled syringes are available in the following strengths:
- 3.0 MIU in 0.5 mL solution
- 9.0 MIU in 0.5 mL solution
On the outer carton, these strengths are expressed as 3.0 x 106 IU/0.5 mL, and 9.0 x 106 IU/0.5 mL of interferon alfa-2a, respectively.
Inactive ingredients
Each pre-filled syringe also contains:
- ammonium acetate
- sodium chloride
- benzyl alcohol
- polysorbate 80
- glacial acetic acid
- sodium hydroxide
- water for injections
Distributor
Roferon-A pre-filled syringes are distributed by:
Roche Products Pty Limited
ABN 70 000 132 865
Level 8, 30-34 Hickson Road
Sydney NSW 2000
Medical Enquiries: 1800 233 950
Please check with your pharmacist for the latest Consumer Medicine Information
Australian Registration Numbers
3.0 MIU: AUST R 58594
9.0 MIU: AUST R 58597
This leaflet was prepared on 27 September 2017

 Histological response was measured in 28 out of 60 patients by the Knodell Histology Activity Index (HAI). Histological improvements were defined as a decrease in the inflammation score of at least 2 points. Histology changes indicated that there was no significant difference between treatment groups.
Histological response was measured in 28 out of 60 patients by the Knodell Histology Activity Index (HAI). Histological improvements were defined as a decrease in the inflammation score of at least 2 points. Histology changes indicated that there was no significant difference between treatment groups. Liver biopsies were conducted post-treatment to determine any improvements in histology. Histological improvements were defined as a decrease in the inflammation score of at least 2 points using the Knodell HAI (see Table 3). Fibrosis was graded according to the Metavir system in which a score of 0 indicated the absence of fibrosis and a score of 4 the presence of cirrhosis. Fibrosis was usually moderate, mild or absent in patients treated with Roferon-A with ribavirin combination therapy but usually severe, moderate or mild in patients treated with Roferon-A alone.
Liver biopsies were conducted post-treatment to determine any improvements in histology. Histological improvements were defined as a decrease in the inflammation score of at least 2 points using the Knodell HAI (see Table 3). Fibrosis was graded according to the Metavir system in which a score of 0 indicated the absence of fibrosis and a score of 4 the presence of cirrhosis. Fibrosis was usually moderate, mild or absent in patients treated with Roferon-A with ribavirin combination therapy but usually severe, moderate or mild in patients treated with Roferon-A alone.
 Ninety-seven (97) patients in the Roferon-A arm and 131 patients in the Avastin arm reduced the dose of Roferon-A from 9 MIU to either 6 or 3 MIU three times a week as prespecified in the protocol.
Ninety-seven (97) patients in the Roferon-A arm and 131 patients in the Avastin arm reduced the dose of Roferon-A from 9 MIU to either 6 or 3 MIU three times a week as prespecified in the protocol.
 If severe adverse reactions or laboratory abnormalities develop during Roferon-A with ribavirin combination therapy modify the doses of each product until the adverse reactions abate. If intolerance persists after dose adjustment, discontinuation of ribavirin or both Roferon-A and ribavirin may be necessary (see Table 7).
If severe adverse reactions or laboratory abnormalities develop during Roferon-A with ribavirin combination therapy modify the doses of each product until the adverse reactions abate. If intolerance persists after dose adjustment, discontinuation of ribavirin or both Roferon-A and ribavirin may be necessary (see Table 7).