

SUMMARY CMI
SPRYCEL®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I using SPRYCEL?
SPRYCEL contains the active ingredient dasatinib. SPRYCEL is used to treat adults and children ages 12 months and older with chronic myeloid leukaemia (CML). It is also used to treat adults and children ages 12 months and older who have a particular form of acute lymphoblastic leukaemia (ALL) called Philadelphia chromosome positive or Ph+ ALL.
For more information, see Section 1. Why am I using SPRYCEL? in the full CMI.
2. What should I know before I use SPRYCEL?
Do not use if you have ever had an allergic reaction to dasatinib or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use SPRYCEL? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with SPRYCEL and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use SPRYCEL?
- Your doctor will decide the dose that is most appropriate for you.
- Please follow your doctor's instructions about how and when to take SPRYCEL.
More instructions can be found in Section 4. How do I use SPRYCEL? in the full CMI.
5. What should I know while using SPRYCEL?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using SPRYCEL? in the full CMI.
6. Are there any side effects?
Like all medicines, this medicine can cause side effects, although not everybody gets them. Your doctor will discuss these with you and will explain the risks and benefits of your treatment. Sometimes they may be serious and you may require medical attention.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
FULL CMI
SPRYCEL® (spry-sell)
Active ingredient: dasatinib (duh-sat-in-ib)
Consumer Medicine Information (CMI)
This leaflet provides important information about using SPRYCEL. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using SPRYCEL.
Where to find information in this leaflet:
1. Why am I using SPRYCEL?
2. What should I know before I use SPRYCEL?
3. What if I am taking other medicines?
4. How do I use SPRYCEL?
5. What should I know while using SPRYCEL?
6. Are there any side effects?
7. Product details
1. Why am I using SPRYCEL?
SPRYCEL contains the active ingredient dasatinib. SPRYCEL acts by inhibiting the activity of proteins within the leukaemia cells of patients with CML or Ph+ ALL. These proteins are responsible for the uncontrolled growth of the leukaemia cells. By inhibiting these proteins, SPRYCEL kills the leukaemia cells in the bone marrow and allows normal red cell, white cell and platelet production to resume.
SPRYCEL is used to treat adults and children ages 12 months and older with chronic myeloid leukaemia (CML). It is also used to treat adults and children ages 12 months and older who have a particular form of acute lymphoblastic leukaemia (ALL) called Philadelphia chromosome positive or Ph+ ALL.
Leukaemia is a cancer of immature white blood cells, which grow in the bone marrow. Under normal circumstances, as these white blood cells mature, they enter the blood stream where they fight infection and maintain the body's immune system. In leukaemia, these immature white blood cells multiply in an uncontrolled manner, occupying the bone marrow space and spilling out into the bloodstream. As a consequence, the production of normal red blood cells (oxygen carrying cells), white blood cells (cells which fight infection), and platelets (cells which help blood to clot) is compromised. Therefore patients with leukaemia are at risk of developing serious anaemia, infections and bleeding.
It is intended that SPRYCEL be used in adults with:
- Newly diagnosed Ph+ CML in the chronic phase who have not received any prior therapies, OR
- Ph+ CML across all phases who are no longer benefiting from other therapies for these diseases (resistance) or in patients who experience severe side effects to other therapies (intolerance), OR
- Newly diagnosed Ph+ ALL who have not received any prior therapies, OR
- Ph+ ALL who are no longer benefiting from other therapies for these diseases (resistance) or in patients who experience severe side effects to other therapies (intolerance).
It is intended that SPRYCEL be used in children with:
- Newly diagnosed Ph+ CML in the chronic phase who have not received any prior therapies, OR
- Ph+ CML in the chronic phase who are no longer benefiting from other therapies for these diseases (resistance) or in patients who experience severe side effects to other therapies (intolerance), OR
- Newly diagnosed Ph+ ALL who have not received any prior therapies.
There is no experience with SPRYCEL treatment in children under 1 year of age.
Ask your doctor if you have any questions about why SPRYCEL was prescribed for you or your child.
2. What should I know before I use SPRYCEL?
Warnings
Do not use SPRYCEL if:
- You or your child are allergic to dasatinib, or any of the ingredients listed at the end of this leaflet.
- Always check the ingredients to make sure you can use this medicine.
- If the expiry date printed on the pack has passed. If you or your child takes this medicine after the expiry date has passed, it may not work as well.
- The packaging is torn or shows signs of tampering.
Check with your doctor if you or your child:
- have any other medical conditions, especially the following: problems with your immune system, liver problem, heart problem, lactose intolerant, hepatitis B infection
If you have not told your doctor about any of the above, tell them before you or your child take SPRYCEL.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Do not take SPRYCEL if you are a pregnant woman or intend to become pregnant.
SPRYCEL can cause harm to the unborn baby if it is given to a pregnant woman.
Tell your doctor if you are a sexually active man.
Men who take SPRYCEL are advised to use a condom to avoid pregnancy in their partner.
Tell your doctor if you are breast-feeding or plan to breast-feed.
Women who are taking SPRYCEL should not breast-feed.
Where to get further information
Your doctor is the best person to answer any further questions you may have about SPRYCEL.
Anything your doctor tells you about SPRYCEL should be followed even if it is different from what is in this leaflet.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may be affected by SPRYCEL or they may affect how well SPRYCEL works.
Tell your doctor if you or your child are taking:
- ketoconazole, itraconazole, ritonavir, atazanavir, erythromycin, clarithromycin and grapefruit juice may all increase the level of SPRYCEL in your bloodstream.
- dexamethasone, phenytoin, carbamazepine, rifampicin, and phenobarbitone may all decrease the levels of SPRYCEL in your bloodstream.
- SPRYCEL may alter the blood levels of cyclosporin.
- Blood thinning medicines such as warfarin sodium, aspirin or non steroidal anti-inflammatory drugs (NSAIDs) taken with SPRYCEL may increase the risk of unwanted bleeding.
- Oral diabetes medicines from the glitazone family of drugs may interact with SPRYCEL.
You or your child should avoid taking cimetidine, famotidine, ranitidine or omeprazole while taking SPRYCEL as they all reduce stomach acid, which is necessary for the absorption of SPRYCEL.
Medicines that neutralise stomach acid (antacids), such as aluminium hydroxide, magnesium hydroxide or calcium carbonate may be taken up to 2 hours before or 2 hours after SPRYCEL.
Some medicines may be affected by SPRYCEL, or may affect how well it works. You or your child may need different amounts of your medicine, or you may need to take different medicines. Your doctor will advise you. Your doctor or pharmacist has more information on medicines to be careful with or avoid while taking SPRYCEL.
Know the medicines you take.
Keep a list of you or your child's medicines to show your doctor or pharmacist.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect SPRYCEL.
4. How do I use SPRYCEL?
How much to take
- The usual starting dose for adults is 100 mg once daily (either as one 100 mg tablet or two 50 mg tablets). The entire dose of 100 mg is to be taken at one time either in the morning or the evening.
- Some adult patients may require an alternative starting dose (140 mg daily taken as two 70 mg tablets). The entire dose of 140 mg is to be taken at one time either in the morning or the evening.
- The starting dose for children is determined by body weight. The entire dose prescribed by your doctor should be taken at one time either in the morning of the evening.
- Your doctor will decide the dose that is most appropriate for you. Please follow your doctor's instructions about how and when to take SPRYCEL.
When to take SPRYCEL
- You or your child should take SPRYCEL consistently at the same time each day (either in the morning or in the evening), with or without a meal, as instructed by your doctor.
How to take SPRYCEL
- Swallow your SPRYCEL tablet(s) whole with a glass of water. Do not break, cut, chew or crush the tablet.
- Do not take SPRYCEL with grapefruit or grapefruit juice.
How long to take SPRYCEL
You or your child should not stop taking SPRYCEL, or reduce the dose without first talking to your doctor. Depending on you or your child's response and any side effects that may be experienced, your doctor may adjust the dose of SPRYCEL, upward or downward, or may temporarily discontinue the medicine.
If you forget to use SPRYCEL
If you miss a dose of SPRYCEL, take the next scheduled dose at its regular time. Don't make up for a missed dose by doubling up on tablets. Call your doctor or pharmacist if you are not sure what to do.
If you use too much SPRYCEL
If you think that you have used too much SPRYCEL, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using SPRYCEL?
Things you should do
- If you or your child are about to be started on any new medicine, tell your doctor and pharmacist that you are taking SPRYCEL.
- Have any tests recommended by your doctor done as soon as possible. Your doctor may order routine laboratory tests to evaluate your blood counts to see how well SPRYCEL is working.
Call your doctor straight away if you or your child:
- become pregnant while taking SPRYCEL
- experience bleeding or easy bruising, no matter how mild
- develop a fever while taking SPRYCEL
- experience shortness of breath and fatigue while taking SPRYCEL
Remind any doctor, dentist or pharmacist you visit that you are using SPRYCEL.
Things you should not do
- Do not give SPRYCEL to anyone else, even if they have the same condition as you or your child.
- Do not stop taking SPRYCEL, or lower the dosage, without checking with your doctor.
- Do not stop taking SPRYCEL tablets because you or your child are feeling better unless advised to do so by your doctor.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how SPRYCEL affects you.
SPRYCEL has been known to cause dizziness or light-headedness in some people. Make sure that you know how you or your child react to SPRYCEL before you drive a car, operate machinery, or do anything else that could be dangerous if you are dizzy.
Looking after your medicine
- Keep SPRYCEL tablets in the pack until it is time to take them. If you or your child take the tablets out of the bottle they may not keep well.
- Keep the pack in a cool dry place where the temperature stays below 30°C.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep this medicine out of the sight and reach of children.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Serious side effects
| Serious side effects | What to do |
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Other side effects
| Common side effects | What to do |
| Speak to your doctor if you have any of these side effects and they worry you. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What SPRYCEL contains
| Active ingredient (main ingredient) | dasatinib |
| Other ingredients (inactive ingredients) |
|
| Potential allergens | lactose monohydrate |
Do not take this medicine if you are allergic to any of these ingredients.
What SPRYCEL looks like
SPRYCEL 20 mg tablet
White to off-white, biconvex, round tablet with "BMS" debossed on one side and "527" on the other
SPRYCEL 50 mg tablet
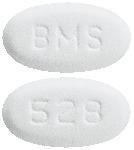
White to off-white, biconvex, oval tablet with "BMS" debossed on one side and "528" on the other
SPRYCEL 70 mg tablet
White to off-white, biconvex, round tablet with "BMS" debossed on one side and "524" on the other
SPRYCEL 100 mg tablet
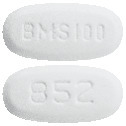
White to off-white, biconvex, oval film-coated tablet with "BMS 100" debossed on one side and "852" on the other
Registration numbers
AUST R 125557 - 20 mg tablet - 60 tablets per bottle
AUST R 125559 - 50 mg tablet - 60 tablets per bottle
AUST R 125561 - 70 mg tablet - 60 tablets per bottle
AUST R 157352 - 100 mg tablet - 30 tablets per bottle
AUST R 125558 - 20 mg tablet - 60 tablets per blister pack (not marketed)
AUST R 125560 - 50 mg tablet - 60 tablets per blister pack (not marketed)
AUST R 125562 - 70 mg tablet - 60 tablets per blister pack (not marketed)
AUST R 157356 - 100 mg tablet - 30 tablets per blister pack (not marketed)
Who distributes SPRYCEL
Bristol-Myers Squibb Australia Pty Ltd
4 Nexus Court, Mulgrave,
Victoria 3170, Australia.
Toll free number: 1800 067 567
Email: [email protected]
SPRYCEL® is a registered trademark of Bristol-Myers Squibb Company
This leaflet was prepared in October 2023.
Published by MIMS December 2023

 Dose escalation is not recommended for paediatric patients with Ph+ ALL, as Sprycel is administered in combination with chemotherapy in these patients.
Dose escalation is not recommended for paediatric patients with Ph+ ALL, as Sprycel is administered in combination with chemotherapy in these patients.
 For paediatric patients with chronic phase CML, if grade ≥ 3 neutropenia or thrombocytopenia recurs during complete haematologic response (CHR), Sprycel should be interrupted, and may be subsequently resumed at a reduced dose. Temporary dose reductions for intermediate degrees of cytopenia and disease response should be implemented as needed.
For paediatric patients with chronic phase CML, if grade ≥ 3 neutropenia or thrombocytopenia recurs during complete haematologic response (CHR), Sprycel should be interrupted, and may be subsequently resumed at a reduced dose. Temporary dose reductions for intermediate degrees of cytopenia and disease response should be implemented as needed.
 Myelosuppression was commonly reported in all patient populations. In newly diagnosed chronic phase CML, myelosuppression was less frequently reported than in chronic phase CML patients with resistance or intolerance to prior imatinib therapy. The frequency of grade 3 or 4 neutropenia, thrombocytopenia, and anaemia was higher in patients with advanced CML or Ph+ ALL than in chronic phase CML.
Myelosuppression was commonly reported in all patient populations. In newly diagnosed chronic phase CML, myelosuppression was less frequently reported than in chronic phase CML patients with resistance or intolerance to prior imatinib therapy. The frequency of grade 3 or 4 neutropenia, thrombocytopenia, and anaemia was higher in patients with advanced CML or Ph+ ALL than in chronic phase CML.
 For time to cCCyR, a hazard ratio of 1.55 indicates that a patient treated with Sprycel is 55% more likely to achieve a cCCyR at any time compared to a patient treated with imatinib. Similarly, for time to MMR, a hazard ratio of 2.01 indicates a patient treated with Sprycel is more than two times more likely to achieve a MMR at any time compared to a patient treated with imatinib. For durability of cCCyR (time in response), a hazard ratio of 0.7 indicates a patient treated with Sprycel is 30% less likely to have disease progression after achieving a cCCyR (or never achieving a cCCyR) compared to a patient treated with imatinib.
For time to cCCyR, a hazard ratio of 1.55 indicates that a patient treated with Sprycel is 55% more likely to achieve a cCCyR at any time compared to a patient treated with imatinib. Similarly, for time to MMR, a hazard ratio of 2.01 indicates a patient treated with Sprycel is more than two times more likely to achieve a MMR at any time compared to a patient treated with imatinib. For durability of cCCyR (time in response), a hazard ratio of 0.7 indicates a patient treated with Sprycel is 30% less likely to have disease progression after achieving a cCCyR (or never achieving a cCCyR) compared to a patient treated with imatinib.
 The progression free survival rate by specific timepoint is displayed graphically in Figure 1. Rate of PFS was consistently higher in Sprycel treated patients who achieved BCR-ABL level ≤ 10 percent at 3 months than those who did not.
The progression free survival rate by specific timepoint is displayed graphically in Figure 1. Rate of PFS was consistently higher in Sprycel treated patients who achieved BCR-ABL level ≤ 10 percent at 3 months than those who did not. The overall survival rate by specific timepoint is displayed graphically in Figure 2. Rate of OS was consistently higher in Sprycel treated patients who achieved BCR-ABL level ≤ 10 percent at 3 months than those who did not.
The overall survival rate by specific timepoint is displayed graphically in Figure 2. Rate of OS was consistently higher in Sprycel treated patients who achieved BCR-ABL level ≤ 10 percent at 3 months than those who did not. The time to MMR is displayed graphically in Figure 3. The time to MMR was consistently shorter in Sprycel treated subjects compared with imatinib treated subjects.
The time to MMR is displayed graphically in Figure 3. The time to MMR was consistently shorter in Sprycel treated subjects compared with imatinib treated subjects. MMR rates by specific timepoint are displayed graphically in Figure 4. Rates of MMR were consistently higher in Sprycel treated subjects compared with imatinib treated subjects.
MMR rates by specific timepoint are displayed graphically in Figure 4. Rates of MMR were consistently higher in Sprycel treated subjects compared with imatinib treated subjects. MR4.5 rates over time are displayed graphically in Figure 5. Rate of MR4.5 over time was consistently higher in Sprycel treated subjects compared with imatinib treated subjects.
MR4.5 rates over time are displayed graphically in Figure 5. Rate of MR4.5 over time was consistently higher in Sprycel treated subjects compared with imatinib treated subjects. Disease progression was defined as increasing white blood cells despite appropriate therapeutic management, loss of CHR (complete haematologic response), partial CyR or CCyR, progression to accelerated phase or blast phase, or death. The estimated 60 month PFS rate was 88.9% (CI: 84.0%-92.4%) and 89.2% (CI: 84.3%-92.7%) for the Sprycel and imatinib treatment groups, respectively. Transformation to accelerated or blast phase occurred less frequently with Sprycel (n = 8; 3.1%) than with imatinib treated patients (n = 15; 5.8%). The estimated 60 month survival rates for Sprycel and imatinib treated patients were 90.9% (CI: 86.6%-93.8%) and 89.6% (CI: 85.2%-92.8%), respectively.
Disease progression was defined as increasing white blood cells despite appropriate therapeutic management, loss of CHR (complete haematologic response), partial CyR or CCyR, progression to accelerated phase or blast phase, or death. The estimated 60 month PFS rate was 88.9% (CI: 84.0%-92.4%) and 89.2% (CI: 84.3%-92.7%) for the Sprycel and imatinib treatment groups, respectively. Transformation to accelerated or blast phase occurred less frequently with Sprycel (n = 8; 3.1%) than with imatinib treated patients (n = 15; 5.8%). The estimated 60 month survival rates for Sprycel and imatinib treated patients were 90.9% (CI: 86.6%-93.8%) and 89.6% (CI: 85.2%-92.8%), respectively. Efficacy was also assessed in patients who were intolerant to imatinib. In this population of patients who received 100 mg once daily, MCyR was achieved in 77% and CCyR in 67% with a minimum of 2 years follow-up.
Efficacy was also assessed in patients who were intolerant to imatinib. In this population of patients who received 100 mg once daily, MCyR was achieved in 77% and CCyR in 67% with a minimum of 2 years follow-up. In the phase III, randomized, open label study in patients with advanced phase CML and Ph+ ALL, whose disease was resistant to or who were intolerant to imatinib, the primary endpoint was MaHR. A total of 611 patients were randomised to either the Sprycel 140 mg once daily or 70 mg twice daily group. Median duration of treatment was approximately 6 months (range < 1-31 months).
In the phase III, randomized, open label study in patients with advanced phase CML and Ph+ ALL, whose disease was resistant to or who were intolerant to imatinib, the primary endpoint was MaHR. A total of 611 patients were randomised to either the Sprycel 140 mg once daily or 70 mg twice daily group. Median duration of treatment was approximately 6 months (range < 1-31 months). In patients with accelerated phase CML treated with the 140 mg once daily regimen, the median duration of MaHR and the median overall survival in patients with accelerated phase CML was not reached for either group; the median PFS was 25 months and 26 months for the 140 mg once daily group and the 70 mg twice daily group, respectively; and the median overall survival was not reached for the 140 mg once daily group and 31 months for the 70 mg twice daily group.
In patients with accelerated phase CML treated with the 140 mg once daily regimen, the median duration of MaHR and the median overall survival in patients with accelerated phase CML was not reached for either group; the median PFS was 25 months and 26 months for the 140 mg once daily group and the 70 mg twice daily group, respectively; and the median overall survival was not reached for the 140 mg once daily group and 31 months for the 70 mg twice daily group. With a median follow-up of 4.5 years in newly diagnosed patients, the median durations of CCyR, MCyR, MMR could not be estimated as more than half of the responding patients had not progressed at the time of data cut-off. Range of duration of response was (2.5+ to 66.5+ months for CCyR), (1.4 to 66.5+ months for MCyR), and (5.4+ to 72.5+ months for subjects who achieved MMR by month 24 and 0.03+ to 72.5+ months for subjects who achieved MMR at any time), where '+' indicates a censored observation.
With a median follow-up of 4.5 years in newly diagnosed patients, the median durations of CCyR, MCyR, MMR could not be estimated as more than half of the responding patients had not progressed at the time of data cut-off. Range of duration of response was (2.5+ to 66.5+ months for CCyR), (1.4 to 66.5+ months for MCyR), and (5.4+ to 72.5+ months for subjects who achieved MMR by month 24 and 0.03+ to 72.5+ months for subjects who achieved MMR at any time), where '+' indicates a censored observation. The chemical name for dasatinib is N-(2-chloro-6-methylphenyl)- 2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl] amino]-5-thiazolecarboxamide, monohydrate.
The chemical name for dasatinib is N-(2-chloro-6-methylphenyl)- 2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl] amino]-5-thiazolecarboxamide, monohydrate.