SUMMARY CMI
TAFINLAR®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
▼ This medicine is new or being used differently. Please report side effects. See the full CMI for further details.
1. Why am I using TAFINLAR?
TAFINLAR contains the active ingredient dabrafenib. In adult patients TAFINLAR is used to treat certain types of skin cancer (melanoma), thyroid cancers and lung cancers; prevent the melanoma coming back if removed by surgery. In children 1 year and older, TAFINLAR is used to treat brain tumours, following other treatments. For more information, see Section 1. Why am I using TAFINLAR? in the full CMI.
2. What should I know before I use TAFINLAR?
Do not use if you have ever had an allergic reaction to dabrafenib or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding. For more information, see Section 2. What should I know before I use TAFINLAR? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with TAFINLAR and affect how it works. A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use TAFINLAR?
- TAFINLAR should be taken at least 1 hour BEFORE eating or 2 hours AFTER eating.
- In adults, the usual total dose of TAFINLAR is 150 mg twice each day (each dose being 12 hours apart).
- TAFINLAR capsules: Take each 150 mg dose on an empty stomach, either as two 75 mg capsules or three 50 mg capsules: one after the other swallowed whole with a full glass of water
- In children, dosage is based on body weight, and is taken twice each day.
- TAFINLAR dispersible tablets are to be taken as a liquid oral solution only. Follow the Instructions for Use in the pack.
More instructions can be found in Section 4. How do I use TAFINLAR? in the full CMI.
5. What should I know while using TAFINLAR?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Drinking alcohol |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using TAFINLAR? in the full CMI.
6. Are there any side effects?
Common side effects include feeling sick, vomiting, headache, changes to appetite, lack of energy, joint pain, hair loss, skin changes including redness, dryness, itching, raised bumps, rash, waxy feel. Serious side effects include difficulty breathing or wheezing, trouble swallowing, swollen hands, feet, or face; chest pain, severe tummy pain, less wee than normal, changes to colour of moles, fever, unexplained bleeding, vision changes. For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
▼ This medicine is subject to additional monitoring due to approval of an extension of indication. This will allow quick identification of new safety information. You can help by reporting any side effects you may get. You can report side effects to your doctor, or directly at www.tga.gov.au/reporting-problems.
FULL CMI
TAFINLAR® (Taf-in-lar)
Active ingredient(s): [dabrafenib (as mesilate)] (da-braf-e-nib mee-sill-ate)
Consumer Medicine Information (CMI)
This leaflet provides important information about using TAFINLAR. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using TAFINLAR.
Where to find information in this leaflet:
1. Why am I using TAFINLAR?
2. What should I know before I use TAFINLAR?
3. What if I am taking other medicines?
4. How do I use TAFINLAR?
5. What should I know while using TAFINLAR?
6. Are there any side effects?
7. Product details
1. Why am I using TAFINLAR?
TAFINLAR contains the active ingredient dabrafenib.
TAFINLAR belongs to a group of medicines called "selective BRAF-inhibitors."
TAFINLAR is used to:
- Treat adults with the following types of cancers that have spread to other parts of the body:
- skin cancers called melanoma
- thyroid cancers called anaplastic thyroid cancer (ATC)
- lung cancers called non-small cell lung cancer (NSCLC) - Prevent melanoma from coming back after the melanoma has been removed by surgery in adult patients.
- Treat children 1 year and older with a brain tumour (glioma), following other treatments.
These cancers have changes (mutations) in a gene called "BRAF" that may have caused your cancer to grow. TAFINLAR slows down or stops the growth of your cancer.
TAFINLAR can be used by itself or together with another medicine called MEKINIST (containing trametinib).
If you are taking these medicines together, please read the MEKINIST Consumer Medicine Information as well as this one carefully.
2. What should I know before I use TAFINLAR?
Monitoring
Before you take TAFINLAR, your doctor will take tumour tissue samples to check whether TAFINLAR is suitable for you.
Your doctor may also take blood samples to monitor your liver while you are taking TAFINLAR.
Warnings
Do not use TAFINLAR if:
- You are pregnant
- You are allergic to "dabrafenib", or any other ingredient listed at the end of this leaflet (symptoms of an allergic reaction can include difficulty breathing, wheezing, swelling of the face, lips or tongue and rash.
- You are under 12 months of age. It is not known whether it is safe and effective in this younger group of patients.
Check with your doctor if you have:
- Liver problems
- Kidney problems
- Diabetes or high levels of sugar in your blood
- Heart problems
- Eye problems
- Lung or breathing problems
- Skin problems
Pregnancy and breastfeeding
- If you are pregnant or breast-feeding, think you may become pregnant or are planning to have a baby, ask your doctor, pharmacist, or health care provider for advice before taking this medicine.
- If you do become pregnant while you are taking TAFINLAR, tell your doctor immediately.
- Your doctor will discuss with you the potential risk(s) of taking TAFINLAR during pregnancy or breast-feeding. TAFINLAR alone or in combination with MEKINIST are not recommended during pregnancy or while breastfeeding.
Birth control requirements
Women taking TAFINLAR
If you are a woman who could become pregnant, you must use good birth control while you are taking TAFINLAR and for:
- at least two weeks after you stop taking it
OR - at least 16 weeks following the last dose of MEKINIST (when taking in combination with TAFINLAR)
Birth control methods containing hormones (such as pills, injections or patches) may not work as well while you are taking TAFINLAR.
- You need to use another good method of birth control, so you don't become pregnant while you are taking TAFINLAR.
- Ask your doctor, healthcare provider, pharmacist, or nurse about options for good birth control or for advice.
Men taking TAFINLAR
You may have a lower sperm count while you are taking TAFINLAR. Your sperm count may not return to normal levels after you stop taking TAFINLAR.
Male patients (including those that have had a vasectomy) with female partners who are or may become pregnant, should use condoms during sexual intercourse while taking TAFINLAR and for at least 2 weeks after stopping TAFINLAR.
If taking TAFINLAR in combination with MEKINIST, male patients should use condoms during sexual intercourse while on TAFINLAR and for at least 16 weeks after stopping both TAFINLAR and MEKINIST.
If you have any further questions on the effect of this medicine on sperm count, ask your doctor or nurse.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins, or supplements that you buy without a prescription from your pharmacy, supermarket, or health food shop.
Some medicines may interfere with TAFINLAR and affect how it works such as:
- Birth control medicines that use hormones
- Warfarin (a medicine used to thin blood and prevent clots)
- Medicines to treat fungal infections (such as ketoconazole, itraconazole, voriconazole, posaconazole)
- Antibiotics such as clarithromycin, telithromycin or rifampicin
- Medicines that cause a low immune system
- Medicines that decrease the amount of stomach acid in your stomach (for example: omeprazole, esomeprazole, rabeprazole, pantoprazole, lansoprazole, ranitidine, cimetidine, famotidine, nizatidine or common antacids.
- Medicines that lower fats in the body such as gemfibrozil
- Medicines that decrease swelling such as dexamethasone
- Medicines used to treat HIV such as ritonavir, saquinavir and atazanavir
- Medicines used to treat seizures phenytoin, phenobarbital, or carbamazepine
- Some anti-depressant medicines such as nefazodone and the herbal St John's wort (Hypericum perforatum)
- Medicines used to treat high levels of cholesterol such as rosuvastatin.
4. How do I use TAFINLAR?
How much to take
Adults
- The usual total dose of TAFINLAR is 150 mg twice each day.
- Follow the instructions provided and use TAFINLAR until your doctor tells you to stop.
You must take 2 TAFINLAR doses each day.
Take each 150 mg dose on an empty stomach as either:
- Two 75 mg capsules, or
- Three 50 mg capsules.
Children (1 year and above)
- The daily dose is based on the body weight and will be determined by their doctor.
- Follow the instructions provided and use TAFINLAR until your doctor tells you to stop.
Do not take more TAFINLAR than your doctor has recommended.
When to take TAFINLAR
Take the first dose of TAFINLAR in the morning and take the second dose of TAFINLAR in the evening, approximately 12 hours later.
Take the morning and evening doses at about the same time each day.
How to take TAFINLAR CAPSULES
Swallow each capsule whole, with a full glass of water. Take the capsules, one after the other, unless your doctor has advised a lower dose.
Take TAFINLAR on an empty stomach
TAFINLAR should be taken either at least:
- 1 hour BEFORE eating
If taking TAFINLAR BEFORE something to eat or drink, take it and then wait at least 1 (one) hour before having any food or drink
OR - 2 hours AFTER eating
If taking TAFINLAR AFTER eating a meal or having a drink, wait at least two (2) hours before taking TAFINLAR.
How to take TAFINLAR DISPERSIBLE TABLETS
TAFINLAR dispersible tablets are to be taken as a liquid oral suspension only and should not be swallowed whole, chewed, or crushed.
If you are taking TAFINLAR dispersible tablets, please follow Instructions for Use in the carton on how to prepare and take TAFINLAR dispersible tablets. Talk to your doctor or pharmacist if you are not sure.
Give TAFINLAR on an empty stomach
TAFINLAR should be given either at least:
- 1 hour BEFORE eating
If taking TAFINLAR BEFORE something to eat or drink, give it and then wait at least 1 (one) hour before having any food or drink
OR - 2 hours AFTER eating
If giving TAFINLAR AFTER eating a meal or having a drink, wait at least two (2) hours before giving TAFINLAR
If you forget to use TAFINLAR
TAFINLAR should be used regularly at the same time each day. If the missed dose of TAFINLAR is:
- Less than 6 hours late, take it as soon as you remember.
- More than 6 hours late, skip that dose and take your next dose at the usual time.
Do not take a double dose to make up for the dose that you missed.
If you use too much
If you think that you have used too much TAFINLAR (or MEKINIST, if taking them together), you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using TAFINLAR?
Things you should do
- Use additional contraception for both men and women to avoid pregnancy while taking TAFINLAR and do this for 2 weeks after stopping TAFINLAR or 16 weeks after stopping MEKINIST and TAFINLAR (if using both medicines together)
- If you become pregnant while taking TAFINLAR, tell your doctor immediately
- Keep all your doctor's appointments so that your progress can be checked
- Have the blood tests your doctor tells you to have
- Have your eyes checked regularly.
Call your doctor straight away if you:
- Get a high fever
- Signs of a fever may include: a temperature of 38°C or more, severe chills, shivering, thirst or dehydration, feeling dizzy, faint, or as if you're going to be sick.
Things you should not do
- Do not stop taking this medicine suddenly.
- Do not take TAFINLAR with food.
Additional Tests
- Your doctor may do some blood tests such as checking blood cells (white blood cells, red blood cells and platelets) and electrolytes (e.g. sodium, phosphorus) in your body from time to time to make sure TAFINLAR is working and to prevent unwanted side effects. Your eyes will also be checked regularly.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how TAFINLAR affects you.
Drinking alcohol
Tell your doctor if you drink alcohol.
Looking after your medicine
- Keep your capsules and dispersible tablets in the bottle until it is time to take them
- Store below 30°C (capsules). Store below 25°C (dispersible tablets).
- Do not remove the desiccant from the capsule bottle or canisters from the dispersible tablet bottle (material that keeps the medicine dry).
- Once dispersible tablets are dispersed, solution should be drunk within 30 minutes.
Follow the instructions in the carton on how to take care of your medicine properly.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep it where young children cannot reach it.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
| TAFINLAR alone Hair problems
Skin problems
| Speak to your doctor if you have any of these less serious side effects and they worry you. If these side effects become severe, please tell your doctor, pharmacist or healthcare provider. |
Serious side effects
| Serious side effects | What to do |
| TAFINLAR alone Skin problems
Signs of infection
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What TAFINLAR contains
| Active ingredient (main ingredient) | Dabrafenib (as mesilate) |
| Other ingredients - capsules (inactive ingredients) | Cellulose - microcrystalline (E460) Magnesium stearate (E572) Silica - colloidal anhydrous Iron oxide red (E172) Titanium dioxide (E171) Hypromellose (E464) Iron oxide black (E172) Shellac Butan-1-ol Isopropyl alcohol Propylene glycol (E1520) Ammonium hydroxide (E527) |
| Potential allergens | This medicine does not contain lactose, sucrose, gluten, tartrazine or any other azo dyes. |
| Other ingredients - dispersible tablets (inactive ingredients) | Mannitol Microcrystalline cellulose Crospovidone Hypromellose Acesulfame potassium agnesium stearate Colloidal anhydrous silica Artificial berry flavour |
| Potential allergens | This medicine does not contain lactose, gluten, tartrazine or any other azo dyes. |
Do not take this medicine if you are allergic to any of these ingredients.
What TAFINLAR looks like
TAFINLAR capsules are available in plastic bottles containing 120 capsules. The bottle has a child resistant closure.
TAFINLAR dispersible tablets are available in plastic bottles containing 210 dispersible tablets. The bottle has a child resistant closure.
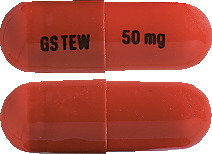
TAFINLAR 50 mg capsules: AUST R 200922
TAFINLAR 50 mg capsules are opaque, hard capsules composed of a dark red body and dark red cap containing a white to slightly coloured solid. The capsule shells are printed with GS TEW and 50 mg.
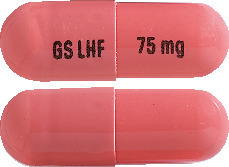
TAFINLAR 75 mg capsules: AUST R 200936
TAFINLAR 75 mg capsules are opaque, hard capsules composed of a dark pink body and dark pink cap containing a white to slightly coloured solid. The capsule shells are printed with GS LHF and 75 mg.
TAFINLAR 10 mg dispersible tablets: AUST R 397093
TAFINLAR 10 mg dispersible tablets are white to slightly yellow, round, biconvex tablets marked "D" on one side and "NVR" on the other.
Who distributes TAFINLAR
Novartis Pharmaceuticals Australia Pty Limited
ABN 18 004 244 160
54 Waterloo Road,
Macquarie Park NSW 2113
Australia
Telephone 1 800 671 203
www.novartis.com.au
® Registered Trademark
This leaflet was prepared in December 2023.
Internal document code: taf291123c based on PI taf291123i.
Published by MIMS January 2024
 The recommended dose and dose level reductions of Tafinlar capsules in paediatric patients who weigh at least 26 kg, is based on body weight (see Table 2). A recommended dose of Tafinlar capsules for patients who weigh less than 26 kg has not been established.
The recommended dose and dose level reductions of Tafinlar capsules in paediatric patients who weigh at least 26 kg, is based on body weight (see Table 2). A recommended dose of Tafinlar capsules for patients who weigh less than 26 kg has not been established. The recommended dosage and dose level reductions for Tafinlar dispersible tablets are based on body weight (see Table 3).
The recommended dosage and dose level reductions for Tafinlar dispersible tablets are based on body weight (see Table 3).


 Table 7 lists the very common (≥ 10% of patients) adverse events reported in the Phase III randomised, open-label study [BREAK-3].
Table 7 lists the very common (≥ 10% of patients) adverse events reported in the Phase III randomised, open-label study [BREAK-3]. See Table 8.
See Table 8.









 In juvenile toxicity studies in rats, effects on growth (shorter long bone length), renal toxicity (tubular deposits, increased incidence of cortical cysts and tubular basophilia and reversible increases in urea and/or creatinine concentrations), thymus toxicity (lymphoid apoptosis) and testicular toxicity (degeneration and tubular dilation) were observed.
In juvenile toxicity studies in rats, effects on growth (shorter long bone length), renal toxicity (tubular deposits, increased incidence of cortical cysts and tubular basophilia and reversible increases in urea and/or creatinine concentrations), thymus toxicity (lymphoid apoptosis) and testicular toxicity (degeneration and tubular dilation) were observed.








 Based on updated data with an additional 29 months of follow-up compared to the primary analysis (minimum follow-up of 59 months), the RFS benefit was maintained with an estimated HR of 0.51 ([95% CI: (0.42, 0.61)] (Figure 5). The 5-year RFS rate was 52% (95% CI: 48, 58) in the combination arm compared to 36% (95% CI: 32, 41) in the placebo arm.
Based on updated data with an additional 29 months of follow-up compared to the primary analysis (minimum follow-up of 59 months), the RFS benefit was maintained with an estimated HR of 0.51 ([95% CI: (0.42, 0.61)] (Figure 5). The 5-year RFS rate was 52% (95% CI: 48, 58) in the combination arm compared to 36% (95% CI: 32, 41) in the placebo arm. Based on 153 events (60 (14%) in the combination arm and 93 (22%) in the placebo arm) corresponding to a 26% information fraction of the total target of 597 OS events, the estimated hazard ratio for OS was 0.57 (95% CI: 0.42, 0.79; p=0.0006). These results did not meet the pre-specified boundary to claim statistical significance at this first OS interim analysis (HR=0.50; p=0.000019). Survival estimates at 1 and 2 years from randomisation were 97% and 91% in the combination arm and 94% and 83% in the placebo arm, respectively. The Kaplan-Meier curve for this OS interim analysis is shown in Figure 6.
Based on 153 events (60 (14%) in the combination arm and 93 (22%) in the placebo arm) corresponding to a 26% information fraction of the total target of 597 OS events, the estimated hazard ratio for OS was 0.57 (95% CI: 0.42, 0.79; p=0.0006). These results did not meet the pre-specified boundary to claim statistical significance at this first OS interim analysis (HR=0.50; p=0.000019). Survival estimates at 1 and 2 years from randomisation were 97% and 91% in the combination arm and 94% and 83% in the placebo arm, respectively. The Kaplan-Meier curve for this OS interim analysis is shown in Figure 6.





