SUMMARY CMI
ZEFFIX
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I using ZEFFIX?
ZEFFIX contains the active ingredient lamivudine. ZEFFIX is used to treat patients 2 years of age or over with long term (chronic) viral infection of the liver caused by hepatitis B.
For more information, see Section 1. Why am I using ZEFFIX? in the full CMI.
2. What should I know before I use ZEFFIX?
Do not use if you have ever had an allergic reaction to lamivudine or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use ZEFFIX? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with ZEFFIX and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use ZEFFIX?
- The normal dose for adults and children aged 12 years or above is one 100 mg tablet once a day.
- Swallow the tablet whole with a glass of water.
More instructions can be found in Section 4. How do I use ZEFFIX? in the full CMI.
5. What should I know while using ZEFFIX?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using ZEFFIX? in the full CMI.
6. Are there any side effects?
Common side effects: headache, vomiting and diarrhoea, nausea, fatigue/ tiredness, abdominal discomfort and pain, coughing with phlegm not associated with cold and flu, dizziness, abnormal liver function tests, muscle disorder (including muscle pain and cramps).
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
FULL CMI
ZEFFIX
Active ingredient(s): lamivudine
Consumer Medicine Information (CMI)
This leaflet provides important information about using ZEFFIX. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using ZEFFIX.
Where to find information in this leaflet:
1. Why am I using ZEFFIX?
2. What should I know before I use ZEFFIX?
3. What if I am taking other medicines?
4. How do I use ZEFFIX?
5. What should I know while using ZEFFIX?
6. Are there any side effects?
7. Product details
1. Why am I using ZEFFIX?
ZEFFIX contains the active ingredient lamivudine. ZEFFIX belongs to a group of medicines called antivirals.
ZEFFIX is used to treat patients 2 years of age or over with long term (chronic) viral infection of the liver caused by hepatitis B.
Hepatitis B is a virus which damages the liver. Treatment with ZEFFIX can reduce the amount of hepatitis B virus in your body. This should lead to less liver damage.
ZEFFIX has not been shown to reduce the risk of passing hepatitis B to others. You will still be able to pass on the hepatitis virus by sexual contact or through your blood. You should use appropriate precautions.
Your doctor may have prescribed ZEFFIX for another reason.
ZEFFIX is not addictive.
If you are already taking this medicine lamivudine for HIV infection (3TC®, Combivir®), your doctor will continue to treat you with the higher dose for that treatment.
2. What should I know before I use ZEFFIX?
Warnings
Do not use ZEFFIX if:
- you are allergic to lamivudine, or any of the ingredients listed at the end of this leaflet.
- Always check the ingredients to make sure you can use this medicine.
Check with your doctor if you:
- have any other medical conditions
- you have, or have ever had, kidney problems
- you have or have ever had problems with your pancreas.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant. The safety of ZEFFIX has not been established in human pregnancy. Use in pregnancy should be considered only if the benefit outweighs the risk.
Talk to your doctor if you are breastfeeding or intend to breastfeed. The safety of ZEFFIX has not been established in breast fed infants. ZEFFIX should only be used if the expected benefit justifies the potential risk to the baby.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with ZEFFIX and affect how it works. These include:
- zalcitabine, used to treat HIV infection (sometimes called the AIDS virus).
- medicines (usually liquids) containing sorbitol and other sugar alcohols (such as xylitol, mannitol, lactitol or maltitol).
- other medicines containing lamivudine, used to treat HIV infection (sometimes called the AIDS virus).
- emtricitabine used to treat HIV infection or hepatitis B infection
- cladribine, used to treat hairy cell leukaemia.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect ZEFFIX.
4. How do I use ZEFFIX?
How much to take
- The normal dose for adults and children aged 12 years or above is one 100 mg tablet once a day.
- Your doctor may prescribe a different dosage, for example if you have kidney problems.
- Follow the instructions provided and use ZEFFIX until your doctor tells you to stop.
When to take / use ZEFFIX
- Your doctor or pharmacist will tell you when to take ZEFFIX each day.
In order for ZEFFIX tablets to be effective, you must take ZEFFIX tablets every day for as long as your doctor says you should take them.
How to take ZEFFIX
- Swallow the tablet whole with a glass of water
- ZEFFIX can be taken with or without food
If you forget to use ZEFFIX
ZEFFIX should be used regularly at the same time each day.
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Do not take a double dose to make up for the dose you missed.
If you use too much ZEFFIX
If you think that you have used too much ZEFFIX, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using ZEFFIX?
Things you should do
Call your doctor straight away if you:
- Become pregnant or intend to become pregnant
- Have not taken ZEFFIX as intended
Remind any doctor, dentist or pharmacist you visit that you are using ZEFFIX.
Things you should not do
- Do not stop using this medicine suddenly or change the dose without first checking with your doctor. If you stop taking ZEFFIX your hepatitis may get worse.
This can cause serious illness particularly if your liver is already not working very well. If you do have to stop taking ZEFFIX your doctor is likely to arrange tests over the following four months to check how well your liver is working.
Each patient responds to the treatment differently. Your doctor will check you by taking regular blood samples. The results will help decide when you should stop taking ZEFFIX.
- Do not give this medicine to anyone else, even if their symptoms seem similar to yours.
- Do not use ZEFFIX to treat any other complaints unless your doctor says to.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how ZEFFIX affects you.
ZEFFIX may cause dizziness and tiredness in some people.
Looking after your medicine
- Store below 30°C.
Follow the instructions in the carton on how to take care of your medicine properly.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep it where young children cannot reach it.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
Serious side effects
| Serious side effects | What to do |
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What ZEFFIX contains
| Active ingredient (main ingredient) | Lamivudine |
| Other ingredients (inactive ingredients) | Microcrystalline cellulose Magnesium stearate Titanium dioxide Hypromellose Sodium starch glycollate Polyethylene glycol Polysorbate 80 Macrogol 400 Iron oxide yellow CI 77192 Iron oxide red CI 77491 |
| Potential allergens | n/a |
Do not take this medicine if you are allergic to any of these ingredients.
ZEFFIX tablets do not contain gluten or lactose.
What ZEFFIX looks like
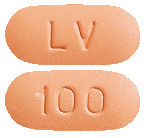
ZEFFIX tablets are butterscotch coloured, film-coated, capsule shaped, biconvex tablets engraved "LV" on one side and "100" on the other.
ZEFFIX tablets are supplied in foil blisters and are in a carton containing 28 tablets.
(AUST R 66828).
Who distributes ZEFFIX
Arrow Pharma Pty Ltd
15-17 Chapel Street
Cremorne VIC 3121
Australia.
This leaflet was prepared in September 2023.
Published by MIMS October 2023

 Data available in patients undergoing intermittent haemodialysis (4 hrs dialysis 2-3 times weekly), indicate that following the initial dosage reduction of Zeffix to correct for the patient's creatinine clearance, no further dosage adjustments are required while undergoing dialysis.
Data available in patients undergoing intermittent haemodialysis (4 hrs dialysis 2-3 times weekly), indicate that following the initial dosage reduction of Zeffix to correct for the patient's creatinine clearance, no further dosage adjustments are required while undergoing dialysis.
 Adverse reactions are listed below by system organ class and frequency. Frequency categories are only assigned to those adverse reactions considered to be at least possibly causally related to lamivudine. Frequencies are defined as: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1000 to < 1/100), rare (≥ 1/10,000 to < 1/1000) and very rare (< 1/10,000).
Adverse reactions are listed below by system organ class and frequency. Frequency categories are only assigned to those adverse reactions considered to be at least possibly causally related to lamivudine. Frequencies are defined as: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1000 to < 1/100), rare (≥ 1/10,000 to < 1/1000) and very rare (< 1/10,000). The molecular formula of lamivudine is C8H11N3O3S and it has a relative molecular mass of 229.3.
The molecular formula of lamivudine is C8H11N3O3S and it has a relative molecular mass of 229.3.