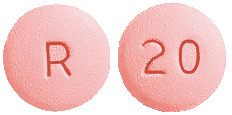
What is in this leaflet
This leaflet answers some of the common questions people ask about CAVSTAT. It does not contain all the information that is known about CAVSTAT.
It does not take the place of talking to your doctor and pharmacist. All medicines have risks and benefits. Your doctor will have weighed the risks of you taking CAVSTAT against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What CAVSTAT is for
CAVSTAT is used to lower high cholesterol levels.
Even though you may have normal cholesterol, CAVSTAT can also be used to reduce the risk of you having a stroke or heart attack if you are a man 50 or more years old or a women 60 or more years old and have at least 2 risk factors for having a heart attack or stroke, such as high blood pressure, low levels of good cholesterol (HDL), smoking or a family history of premature coronary heart disease. Your doctor may also do a blood test to measure a substance called C Reactive Protein to help decide if you should be given CAVSTAT for this use.
Cholesterol and Triglycerides
Everyone has cholesterol and triglycerides in their blood. They are fatty substances needed by the body for many things.
Triglycerides are an energy source for the body. Cholesterol is used for such things as building cells, making bile acids (which help to digest foods) and making some hormones.
There are different types of cholesterol. Too much of the "bad" cholesterol (LDL) can block the blood vessels that supply your heart and brain with blood, and can cause heart attack, angina and stroke. The "good" cholesterol (HDL) helps to remove the bad cholesterol from the blood vessels. High levels of triglycerides can be associated with a low level of "good" cholesterol and may increase the risk of heart disease.
How CAVSTAT works
CAVSTAT belongs to a group of medicines known as HMG-CoA reductase inhibitors (also known as 'statins'). It lowers the "bad" cholesterol, and raises the "good" cholesterol when exercise and changes to diet are not enough on their own.
Cholesterol is present in many foods and is also made by your body. CAVSTAT does not reduce the cholesterol that comes from fat in food. Because of this, when you are taking CAVSTAT, you need to follow a low-fat diet, control your weight and exercise regularly.
High cholesterol is also more likely to occur with certain diseases or if you have a family history of high cholesterol.
Your doctor will have explained why you are being treated with CAVSTAT and told you what dose to take. Your doctor may need to check your cholesterol levels before prescribing CAVSTAT or changing your dose.
Follow all directions given to you by your doctor carefully. They may differ from the information contained in this leaflet. Your doctor may prescribe this medicine for another use. Ask your doctor if you want more information.
CAVSTAT is not addictive.
CAVSTAT is not recommended for use in children as its effects in children have not been established.
Before you take CAVSTAT
When you must not use it
Do not use CAVSTAT if you are pregnant or intend to become pregnant. Ask your doctor about effective methods of contraception.
If you become pregnant, stop taking CAVSTAT as soon as you find out and see your doctor immediately.
Do not use CAVSTAT if you are breast feeding. We do not know if your baby can take in CAVSTAT from breast milk if you are breast feeding.
Do not use CAVSTAT if you have active liver disease or if tests show you have elevated levels of liver enzymes which may show that you have a problem with your liver.
Do not use CAVSTAT 40 mg if you have:
- low thyroid hormone levels (hypothyroidism)
- a personal or family history of hereditary muscular disorders
- a previous history of muscular problems from using other lipid-lowering agents
- a history of very heavy alcohol use
- Asian heritage
- been prescribed another class of lipid lowering agent called a fibrate
- been prescribed any medicine containing fusidic acid
- severe kidney impairment
- situations that may increase rosuvastatin blood levels
Do not use after the use by (expiry) date printed on the pack. It may have no effect at all, or worse, an entirely unexpected effect if you take it after the expiry date.
Do not use CAVSTAT if the packaging is torn or shows signs of tampering.
Do not use it to treat any other complaints unless your doctor tells you to. Do not give this medicine to anyone else.
Before you start to use it
You must tell your doctor if:
- you have any allergies to
- any other statins such as simvastatin (e.g. ZOCOR, LIPEX); pravastatin (e.g. PRAVACHOL); atorvastatin (e.g. LIPITOR); fluvastatin (e.g. VASTIN)
- any ingredients listed at the end of this leaflet
If you have an allergic reaction, you may get a skin rash, hay fever, difficulty in breathing or feel faint. - you have any of these medical conditions
- liver problems
- kidney problems
- low thyroid hormone levels (hypothyroidism)
- a personal or family history of muscle disorders
- a history of muscle problems from using other lipid-lowering agents
It may not be safe for you to take CAVSTAT if you have any of these conditions. Your doctor may do a blood test to check if you have any problems, and may adjust the dose of CAVSTAT. - you have any unexplained aches or pains in your muscles
- you regularly drink large amounts of alcohol
Excessive alcohol consumption may not be safe in patients taking CAVSTAT.
Taking other medicines
Tell your doctor if you are taking any other medicines including
- cyclosporin (e.g. SANDIMMUN and NEORAL, used, for example, after organ transplant)
- antacids (medicines used to treat heartburn and indigestion). CAVSTAT can be taken 2 hours before or 2 hours after taking an antacid.
- warfarin (e.g. COUMADIN and MAREVAN, used to stop blood clots).
- clopidogrel (e.g. PLAVIX), a medicine used to prevent blood clots
- gemfibrozil (e.g. LOPID, JEZIL and AUSGEM, used to lower blood lipids).
- Fusidic acid (e.g. FUCIDIN) used to treat some infections.
- various protease inhibitors used in combination with ritonavir to treat HIV infection (e.g. KALETRA).
- simeprevir (OLYSIO), a medicine used for treatment of chronic hepatitis C
- eltrombopag (REVOLADE), used to increase your platelet count in your blood
- medicines that you buy at the chemist, supermarket or health food shop, including herbal medicines.
Your doctor will consider if CAVSTAT should be used together with any of these medicines, or may wish to adjust the dose of CAVSTAT or the other medicines. These medicines may affect the way CAVSTAT works.
Your doctor or pharmacist can tell you what to do if you are taking any of these medicines.
If you have not told your doctor about any of these things, tell them before you take CAVSTAT.
Effects on driving and using machinery
Be careful driving a car or operating machinery until you know if CAVSTAT affects you.
CAVSTAT generally does not cause any problems with your ability to drive a car or operate machinery. However, as with many other medicines, CAVSTAT may cause dizziness in some people.
Using CAVSTAT
How to take it
Depending on your condition and ethnic background, your doctor will decide the most appropriate starting dose for you.
If you have high cholesterol, your doctor will probably start you on 5 mg or 10 mg tablet taken once daily.
Your doctor will then monitor your cholesterol and triglyceride levels during your treatment, and, if needed, may increase your CAVSTAT dose to 20 mg once daily. For most patients a maximum 20 mg CAVSTAT daily dose is sufficient to treat high cholesterol.
A small number of patients may need to further increase their CAVSTAT dose to 40 mg once daily, for example patients whose high cholesterol is hereditary.
If your cholesterol is not high but you have risks for having a heart attack or stroke, your doctor may start you on 20 mg.
Your doctor will advise you on the dose that's right for your condition. The daily dose of CAVSTAT must not exceed 40 mg daily.
DO NOT INCREASE OR ADJUST YOUR CAVSTAT DOSE YOURSELF.
Take CAVSTAT once a day, at about the same time each day. Keeping a regular time for taking CAVSTAT will help to remind you to take it.
Swallow each tablet whole with a drink of water.
CAVSTAT can be taken at any time of the day. It does not matter whether you take CAVSTAT with food or on an empty stomach.
While taking CAVSTAT you also need to follow a low-fat diet, control your weight and exercise regularly.
How long to take it
You must continue to take it as directed.
CAVSTAT helps lower your blood cholesterol and triglycerides. It does not cure your condition. If you stop taking CAVSTAT, your cholesterol and triglycerides levels may rise again.
You may have to take cholesterol lowering medicines for the rest of your life.
If you forget to take it
If you forget to take a dose of CAVSTAT, take it as soon as you remember, as long as it is more than 6 hours before your next dose is due. Otherwise, wait until your next dose is due and take it as normal.
Do not double the dose to make up for the one you missed.
If you have trouble remembering when to take your medicine, ask your pharmacist for some hints.
Overdose
Telephone your doctor or the Poisons Information Centre (13 11 26), or go to Accident and Emergency at your nearest hospital immediately if you think that you or anyone else may have taken too much CAVSTAT even if there are no signs of discomfort or poisoning.
While you are using CAVSTAT
Things you must do
Have your blood cholesterol and triglycerides checked when your doctor says so to make sure CAVSTAT is working.
Stop taking CAVSTAT and contact your doctor immediately if you become pregnant while you are taking CAVSTAT.
Things you must not do
Do not stop taking CAVSTAT unless you have discussed it with your doctor.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking CAVSTAT.
CAVSTAT helps most people with too much cholesterol, but it may have unwanted side effects in a few people.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice any of the following and they worry you:
- headache
- constipation
- dizziness
- nausea (feeling sick)
- stomach pain
- unusual tiredness
- itchy skin
- memory loss
- stiff or painful joints (arthralgia)
These side effects are usually mild.
Tell your doctor if you notice a significant increase in your need to urinate or if you are significantly more hungry or thirsty than usual.
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you notice any of the following:
- aching muscles, muscle tenderness or weakness not caused by exercise, particularly if you also have a fever or generally feel unwell
- difficulty breathing, swelling of the face, eyelids or lips
- difficulty breathing, coughing, particularly if you also feel generally unwell (e.g. fatigue, weight loss, fever).
These are all serious side effects. You may need urgent medical attention
Serious side effects are rare.
Tell your doctor if you notice anything else that is making you feel unwell. Some people may get other side effects while taking CAVSTAT.
Do not be alarmed by this list of side effects. You may not experience any of them.
After using it
Storage
Keep your tablets in the blister/bottle pack until it is time to take them. If you take CAVSTAT out of the blister/bottle pack it will not keep well.
Keep it in a cool dry place where the temperature stays below 25°C.
Do not store it or any other medicine in the bathroom or near a sink. Heat and dampness can destroy some medicines.
Keep it where young children cannot reach it. A locked cupboard, at least one-and-a-half metres above the ground, is a good place to store medicines.
Do not leave it in the car on hot days.
Disposal
Ask your pharmacist what to do with any CAVSTAT tablets you have left over if your doctor tells you to stop taking them, or you find that the expiry date has passed.
Product description
What CAVSTAT looks like
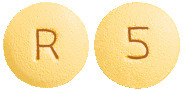
Cavstat film-coated tablets 5 mg are yellow, round, biconvex, film-coated tablet, debossed "5" on one side and "R" on other side.
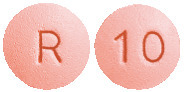
Cavstat film-coated tablets 10 mg are pink, round, biconvex, film-coated tablet, debossed "10" on one side and "R" on other side.
Cavstat film-coated tablets 20 mg are pink, round, biconvex, film-coated tablet, debossed "20" on one side and "R" on other side.
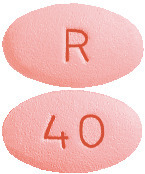
Cavstat film-coated tablets 40 mg are pink, oval, biconvex, film-coated tablet, debossed "40" on one side and "R" on other side.
Ingredients
Cavstat contains rosuvastatin calcium as the active ingredient, equivalent to rosuvastatin 5mg 10mg, 20mg and 40mg
plus
- Lactose
- Lactose anhydrous
- Microcrystalline cellulose
- Light magnesium oxide
- Crospovidone
- Magnesium stearate
- Glycerol triacetate
- Hypromellose
- Titanium dioxide
- Allura red AC aluminium lake
- Brilliant blue FCF aluminium lake.
The 5 mg tablets also contain quinoline yellow whereas the 10 mg, 20 mg and 40 mg tablets contain sunset yellow FCF aluminium lakes.
Name and Address of the Sponsor
Accord Healthcare Pty Ltd
Level 24, 570 Bourke St
Melbourne, VIC, 3000
Australia
Australian Registration Numbers:
5 mg blister pack: AUST R 234543
5 mg bottle pack: AUST R 235277
10 mg blister pack: AUST R 234485
10 mg bottle pack: AUST R 234548
20 mg blister pack: AUST R 234514
20 mg bottle pack: AUST R 234476
40 mg blister pack: AUST R 234508
40 mg bottle pack: AUST R 234461
Date of Approval
September 2016
Published by MIMS April 2017


 The percent change from baseline in HDL-C at week 6 is shown in Figure 2.
The percent change from baseline in HDL-C at week 6 is shown in Figure 2. The mean percent change in HDL-C from baseline to week 6 for each statin treatment group represented in Figure 2 is summarised with 95% CI in Table 3.
The mean percent change in HDL-C from baseline to week 6 for each statin treatment group represented in Figure 2 is summarised with 95% CI in Table 3. Table 4 summarises the pooled lipid variable data for rosuvastatin 5 and 10 mg from 5 phase III efficacy trials (trials 24-28).
Table 4 summarises the pooled lipid variable data for rosuvastatin 5 and 10 mg from 5 phase III efficacy trials (trials 24-28).


 There were no statistically significant reductions in the rate of noncardiovascular death or the incidence of bone fractures in the rosuvastatin treated group compared to placebo.
There were no statistically significant reductions in the rate of noncardiovascular death or the incidence of bone fractures in the rosuvastatin treated group compared to placebo. The individual components of the primary end point are presented in Figure 4. Rosuvastatin significantly reduced the risk of nonfatal myocardial infarction, nonfatal stroke, and arterial revascularization procedures. There were no significant treatment differences between the rosuvastatin and placebo groups for death due to cardiovascular causes or hospitalizations for unstable angina.
The individual components of the primary end point are presented in Figure 4. Rosuvastatin significantly reduced the risk of nonfatal myocardial infarction, nonfatal stroke, and arterial revascularization procedures. There were no significant treatment differences between the rosuvastatin and placebo groups for death due to cardiovascular causes or hospitalizations for unstable angina. In a post hoc subgroup analysis of JUPITER subjects (n = 1405; rosuvastatin = 725, placebo = 680) with a hsCRP ≥ 2 mg/L and no other traditional risk factors (smoking, BP ≥ 140/90 or taking antihypertensives, low HDL-C) other than age, after adjustment for high HDL-C, there was no significant treatment benefit with rosuvastatin treatment.
In a post hoc subgroup analysis of JUPITER subjects (n = 1405; rosuvastatin = 725, placebo = 680) with a hsCRP ≥ 2 mg/L and no other traditional risk factors (smoking, BP ≥ 140/90 or taking antihypertensives, low HDL-C) other than age, after adjustment for high HDL-C, there was no significant treatment benefit with rosuvastatin treatment. The chemical name is bis [(E)-7-[4-(4-fluorophenyl)-6-isopropyl-2-[methyl (methylsulfonyl) amino] pyrimidin-5-yl] (3R, 5S)-3,5-dihydroxyhept-6-enoic acid] calcium salt.
The chemical name is bis [(E)-7-[4-(4-fluorophenyl)-6-isopropyl-2-[methyl (methylsulfonyl) amino] pyrimidin-5-yl] (3R, 5S)-3,5-dihydroxyhept-6-enoic acid] calcium salt.