

WHAT IS IN THIS LEAFLET
This leaflet answers some common questions about Pravastatin Sandoz.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking this medicine against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
WHAT PRAVASTATIN SANDOZ IS USED FOR
This medicine is used to lower high blood cholesterol levels (doctors call this hypercholesterolaemia).
It is also used in people who have had a heart attack or an episode of unstable angina, even when their cholesterol levels are normal.
It is also used to treat heterozygous familial hypercholesterolaemia in children and adolescent patients aged 8 years and older as an adjunct to diet and lifestyle changes.
It contains the active ingredient pravastatin sodium. Pravastatin sodium belongs to a group of medicines called HMG-CoA reductase inhibitors.
It works by reducing the level of cholesterol in your blood and helps to protect you in other ways from heart attack or stroke.
It is more effective if it is taken with a diet low in fat.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is not addictive.
This medicine is available only with a doctor's prescription.
BEFORE YOU TAKE PRAVASTATIN SANDOZ
When you must not take it
Do not take this medicine if you have an allergy to:
- pravastatin sodium, the active ingredient, or to any of the other ingredients listed at the end of this leaflet under Product Description.
- any other similar medicines, especially if they are in the same drug class as Pravastatin Sandoz (HMG-CoA reductase inhibitor).
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
Do not take this medicine if you have or have had problems with your liver.
Do not take this medicine if you are pregnant. It may affect your developing baby if you take it during pregnancy.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- kidney problems
- liver problems. Your doctor will do a blood test to make sure you have no problems with your liver.
- muscle pain from other medicines used to treat high cholesterol
- breathing discomfort, persistent cough, fatigue, weight loss and fever
- suffer from hormonal disorders
- suffer from central nervous system vascular lesions
- suffer from allergies
- suffer from homozygous familial hypercholesterolaemia (a doctor will have told you this)
- have increased triglycerides in your blood (a doctor will have told you this also)
- suffer from muscle disease (including pain, tenderness or weakness).
Tell your doctor if you plan on becoming pregnant or will be breastfeeding while you are using Pravastatin Sandoz. You doctor can discuss with you the risks and benefits involved.
Tell your doctor if you drink alcohol regularly.
If you have not told your doctor about any of the above, tell him/her before you start taking Pravastatin Sandoz.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and Pravastatin Sandoz may interfere with each other. These include:
- any other medicine to lower cholesterol, such as gemfibrozil, cholestyramine or colestipol
- ciclosporin, a medicine used to suppress the immune system
- ketoconazole, a medicine used to treat fungal infections
- spironolactone, a medicine used to treat high blood pressure
- cimetidine, a medicine used to treat ulcers
- gemfibrozil
- cholestyramine and colestipol
- antacids
- macrolides
- propanol
- bile acid sequestrants
- digoxin
- warfarin or other coumarin anticoagulants.
These medicines may be affected by Pravastatin Sandoz or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
It generally does not interfere with your ability to drive or operate machinery. However some people may experience dizziness, so you should be sure how you react to Pravastatin Sandoz before you drive a car, or operate machinery.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
HOW TO TAKE PRAVASTATIN SANDOZ
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions, ask your doctor or pharmacist for help.
How much to take
Adults - The usual dose is 10 to 80 mg per day for lowering cholesterol and 40mg for reducing the risk of a stroke or heart attack.
If you have kidney or liver problems, or if you are over 65 years old, the starting dose is 10mg.
Children (8 to 13 years old) with heterozygous familial hypercholesterolaemia - The usual dose is 20mg once daily.
Adolescents (14 to 18 years old) with heterozygous familial hypercholesterolaemia - The usual dose is 40mg once daily.
Ask your doctor or pharmacist if you are unsure of the correct dose for you. They will tell you exactly how much to take.
Follow the instructions they give you.
If you take the wrong dose, Pravastatin Sandoz may not work as well and your problem may not improve.
How to take it
Swallow the tablets with a glass of water or another liquid. The tablets can be broken in half. If you need to break Pravastatin Sandoz, hold the tablet with both hands and snap along the break line.
Pravastatin Sandoz can be taken with or without food.
When to take Pravastatin Sandoz
Take your medicine once a day in the evening before bedtime.
Taking it at the same time each day will have the best effect. It will also help you to remember when to take it.
How long to take Pravastatin Sandoz
Continue taking your medicine for as long as your doctor tells you.
This medicine helps to control your condition, but does not cure it. It is important to keep taking your medicine even if you feel well.
If you forget to take it
Take your dose as soon as you remember, and continue to take it as you would normally.
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much Pravastatin Sandoz. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
WHILE YOU ARE TAKING PRAVASTATIN SANDOZ
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Pravastatin Sandoz.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you become pregnant while taking this medicine, tell your doctor immediately.
Keep all of your doctor's appointments so that your progress can be checked. Your doctor may do some tests (such as checking your cholesterol levels) from time to time to make sure the medicine is working and to prevent unwanted side effects.
Things you must not do
Do not take Pravastatin Sandoz to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Things to be careful of
Be careful driving or operating machinery until you know how Pravastatin Sandoz affects you. This medicine generally does not cause any problems with your ability to drive a car or operate machinery. However, as with many other medicines, Pravastatin Sandoz may cause dizziness in some people. If you have any of those symptoms, do not drive, operate machinery or do anything else that could be dangerous.
SIDE EFFECTS
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Pravastatin Sandoz.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- upset stomach, nausea, diarrhoea, wind
- constipation
- headache
- dizziness
- blurred vision
- fatigue
- sleep disturbance
- sexual dysfunction
- depression.
Tell your doctor as soon as possible if you notice any of the following:
- unexplained muscle pain, muscle tenderness or weakness, muscle cramps
- chest pain, feeling of tightness, pressure or heaviness in the chest.
The above lists include the most common side effects of the medicine.
Tell your doctor immediately if you notice any of the following:
- swelling of the face, lips, mouth, tongue or throat which may cause difficulty in swallowing or breathing
- joint pain
- skin rash or itchiness
- sensitivity to light
- fever, generally feeling unwell.
These are the symptoms of an hypersensitivity reaction.
In few cases, statins have been reported to induce de novo or aggravate preexisting myasthenia gravis or ocular myasthenia.
Tell your doctor or pharmacist if you notice anything else that is making you feel unwell. Other side effects not listed above may also occur in some people.
AFTER TAKING PRAVASTATIN SANDOZ
Storage
Keep your medicine in the original container.
If you take it out of its original container it may not keep well.
Keep your medicine in a cool dry place where the temperature stays below 25°C.
Do not store Pravastatin Sandoz or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car.
Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
PRODUCT DESCRIPTION
What it looks like
Pravastatin Sandoz comes in four types of tablets:
Pravastatin Sandoz 10 mg - light brown, oval tablet, scored on both sides and marked with "P10".
Pravastatin Sandoz 20 mg - light brown, oval tablet, scored on both sides and marked with "P20".
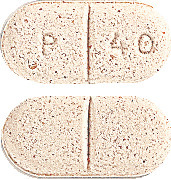
Pravastatin Sandoz 40 mg - light brown, oval tablet, scored on both sides and marked with "P40".
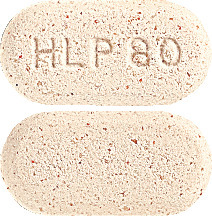
Pravastatin Sandoz 80 mg - light brown, oval tablet, marked with "HLP80".
Available in blister packs of 30 tablets.
Not all strengths may be marketed.
Ingredients
Active ingredient:
- Pravastatin Sandoz 10 mg - 10 mg pravastatin sodium
- Pravastatin Sandoz 20 mg - 20 mg pravastatin sodium
- Pravastatin Sandoz 40 mg - 40 mg pravastatin sodium
- Pravastatin Sandoz 80 mg - 80 mg pravastatin sodium
Inactive ingredients:
- microcrystalline cellulose
- lactose monohydrate
- dibasic sodium phosphate
- croscarmellose sodium
- sodium lauryl sulphate
- povidone
- iron oxide red CI77491
- colloidal anhydrous silica
- magnesium stearate.
This medicine does not contain sucrose, gluten, tartrazine or any other azo dyes.
Supplier
Sandoz Pty Ltd
100 Pacific Highway
North Sydney, NSW 2060
Australia
Tel 1800 726 369
This leaflet was revised in January 2024.
Australian Register Numbers
Pravastatin Sandoz 10 mg tablets: AUST R 152451
Pravastatin Sandoz 20 mg tablets: AUST R 152458
Pravastatin Sandoz 40 mg tablets: AUST R 152450
Pravastatin Sandoz 80 mg tablets: AUST R 152452
® Registered Trade Mark. The trade marks mentioned in this material are the property of their respective owners.
Published by MIMS March 2024

 There was no statistically significant difference in the change in lens opacity between the control and pravastatin treatment groups during this time interval.
There was no statistically significant difference in the change in lens opacity between the control and pravastatin treatment groups during this time interval. In a pooled analysis of two multicenter, double blind, placebo-controlled studies in patients with primary hypercholesterolaemia, treatment with pravastatin at a daily dose of 80 mg increased HDL-C and significantly decreased Total-C, LDL-C, and TG from baseline after 6 weeks. The efficacy results of the individual studies were consistent with the pooled data. Mean percent changes from baseline after 6 weeks of treatment were: Total-C (-27%), LDL-C (-37%), HDL-C (+3%) and TG (-19%), with placebo-subtracted changes for LDL-C and TG of -36% and -20% respectively.
In a pooled analysis of two multicenter, double blind, placebo-controlled studies in patients with primary hypercholesterolaemia, treatment with pravastatin at a daily dose of 80 mg increased HDL-C and significantly decreased Total-C, LDL-C, and TG from baseline after 6 weeks. The efficacy results of the individual studies were consistent with the pooled data. Mean percent changes from baseline after 6 weeks of treatment were: Total-C (-27%), LDL-C (-37%), HDL-C (+3%) and TG (-19%), with placebo-subtracted changes for LDL-C and TG of -36% and -20% respectively. The effect on the combined endpoint of coronary heart disease death or non-fatal myocardial infarction was evident as early as six months after beginning pravastatin therapy.
The effect on the combined endpoint of coronary heart disease death or non-fatal myocardial infarction was evident as early as six months after beginning pravastatin therapy.

 In the Long-term Intervention with Pravastatin in Ischaemic Disease (LIPID) study, the effect of pravastatin 40 mg daily was assessed in 9,014 men and women with normal to elevated serum cholesterol levels (baseline total C = 4.0 to 7.0 mmol/L; mean total C = 5.66 mmol/L; mean total C/HDL C ratio = 5.9), and who had experienced either a myocardial infarction or had been hospitalised for unstable angina pectoris in the preceding 3 to 36 months. Patients with a wide range of baseline levels of triglycerides were included (≤ 5.0 mmol/L) and enrolment was not restricted by baseline levels of HDL cholesterol. At baseline, 82% of patients were receiving aspirin, 76% were receiving antihypertensive medication, and 41% had undergone myocardial revascularisation. Patients in this multicentre, double blind, placebo-controlled study participated for a mean of 5.6 years (median = 5.9 years). Treatment with pravastatin significantly reduced the risk for CHD death by 24% (p = 0.0004). The risk for coronary events (either CHD death or non-fatal myocardial infarction) was significantly reduced by 24% (p < 0.0001) in the pravastatin treated patients. The risk for fatal or non-fatal myocardial infarction was reduced by 29% (p < 0.0001). Pravastatin reduced both the risk for total mortality by 23% (p < 0.0001) and cardiovascular mortality by 25% (p < 0.0001). The risk for undergoing myocardial revascularisation procedures (coronary artery bypass grafting or percutaneous transluminal coronary angioplasty) was significantly reduced by 20% (p < 0.0001) in the pravastatin treated patients. Pravastatin also significantly reduced the risk for stroke by 19% (p = 0.0477). Treatment with pravastatin significantly reduced the number of days of hospitalisation per 100 person-years of follow-up by 15% (p < 0.001). The prespecified subgroup (age, sex, hypertensive, diabetics, smokers, lipid subgroups) analyses were conducted using the combined endpoint of CHD and non-fatal myocardial infarction. The study was not powered to examine results within each subgroup but formal testing for heterogeneity of treatment effect was undertaken across each of the subgroups and no significant heterogeneity was found (p ≥ 0.08), i.e. a consistent treatment effect was seen with pravastatin therapy across all patient subgroups and event parameters. Among patients who qualified with a history of myocardial infarction, pravastatin significantly reduced the risk for total mortality by 25% (p = 0.0016); for CHD mortality by 23% (p = 0.004); for CHD events by 22% (p = 0.002) and for fatal or non-fatal myocardial infarction by 25% (p = 0.0008). Among patients who qualified with a history of hospitalisation for unstable angina pectoris, pravastatin significantly reduced the risk for total mortality by 26% (p = 0.0035); for CHD mortality by 26% (p = 0.0358); for CHD events by 29% (p = 0.0001) and for fatal or non-fatal myocardial infarction by 37% (p = 0.0003). The results of the LIPID study are shown in Table 7.
In the Long-term Intervention with Pravastatin in Ischaemic Disease (LIPID) study, the effect of pravastatin 40 mg daily was assessed in 9,014 men and women with normal to elevated serum cholesterol levels (baseline total C = 4.0 to 7.0 mmol/L; mean total C = 5.66 mmol/L; mean total C/HDL C ratio = 5.9), and who had experienced either a myocardial infarction or had been hospitalised for unstable angina pectoris in the preceding 3 to 36 months. Patients with a wide range of baseline levels of triglycerides were included (≤ 5.0 mmol/L) and enrolment was not restricted by baseline levels of HDL cholesterol. At baseline, 82% of patients were receiving aspirin, 76% were receiving antihypertensive medication, and 41% had undergone myocardial revascularisation. Patients in this multicentre, double blind, placebo-controlled study participated for a mean of 5.6 years (median = 5.9 years). Treatment with pravastatin significantly reduced the risk for CHD death by 24% (p = 0.0004). The risk for coronary events (either CHD death or non-fatal myocardial infarction) was significantly reduced by 24% (p < 0.0001) in the pravastatin treated patients. The risk for fatal or non-fatal myocardial infarction was reduced by 29% (p < 0.0001). Pravastatin reduced both the risk for total mortality by 23% (p < 0.0001) and cardiovascular mortality by 25% (p < 0.0001). The risk for undergoing myocardial revascularisation procedures (coronary artery bypass grafting or percutaneous transluminal coronary angioplasty) was significantly reduced by 20% (p < 0.0001) in the pravastatin treated patients. Pravastatin also significantly reduced the risk for stroke by 19% (p = 0.0477). Treatment with pravastatin significantly reduced the number of days of hospitalisation per 100 person-years of follow-up by 15% (p < 0.001). The prespecified subgroup (age, sex, hypertensive, diabetics, smokers, lipid subgroups) analyses were conducted using the combined endpoint of CHD and non-fatal myocardial infarction. The study was not powered to examine results within each subgroup but formal testing for heterogeneity of treatment effect was undertaken across each of the subgroups and no significant heterogeneity was found (p ≥ 0.08), i.e. a consistent treatment effect was seen with pravastatin therapy across all patient subgroups and event parameters. Among patients who qualified with a history of myocardial infarction, pravastatin significantly reduced the risk for total mortality by 25% (p = 0.0016); for CHD mortality by 23% (p = 0.004); for CHD events by 22% (p = 0.002) and for fatal or non-fatal myocardial infarction by 25% (p = 0.0008). Among patients who qualified with a history of hospitalisation for unstable angina pectoris, pravastatin significantly reduced the risk for total mortality by 26% (p = 0.0035); for CHD mortality by 26% (p = 0.0358); for CHD events by 29% (p = 0.0001) and for fatal or non-fatal myocardial infarction by 37% (p = 0.0003). The results of the LIPID study are shown in Table 7.
 The safety and efficacy of pravastatin doses above 40 mg daily have not been studied in children. The long-term efficacy of pravastatin therapy in childhood to reduce morbidity and mortality in adulthood has not been established.
The safety and efficacy of pravastatin doses above 40 mg daily have not been studied in children. The long-term efficacy of pravastatin therapy in childhood to reduce morbidity and mortality in adulthood has not been established.
