Diagnostic tests
DNA i. approach and techniques
- C.G. Woods, D.M. Danks
- Aust Prescr 1995;18:45-8
- 1 April 1995
- DOI: 10.18773/austprescr.1995.050
Summary
The genes responsible for the thousands of known human genetic diseases are being identified at an increasingly rapid rate. The technological advances which are identifying these genes are also simplifying and speeding up the testing for individual diseases.DNA testing is used to detect mutations when biochemical tests for a disease are not available or when DNA tests are more practical. Individuals and families considered for testing require counselling and practitioners will need to liaise with a clinical genetics service.
Introduction
To date, DNA tests have mainly been used in those diseases where the basic biochemical physiological defect has not been discovered. Outstanding examples are Duchenne muscular dystrophy, cystic fibrosis, Huntington's disease and myotonic dystrophy. In other genetic diseases, especially inborn errors of metabolism, the traditional methods of research have resulted in a precise knowledge of the basic biochemical defects and good diagnostic tests for patients or a fetus at risk. In most of these diseases, the traditional assays are still preferred to DNA testing for prenatal diagnosis. This is because an enzyme assay will give an abnormal result in all cases of a particular inborn error of metabolism, despite the fact that each of the families concerned may have different mutations in the gene. However, DNA tests have become important in some of these diseases because the biochemical disturbance is not expressed in the accessible amniotic cells or chorionic villi. Thus, DNA tests have proved very valuable in thalassaemias and haemophilias, for which fetal blood sampling was previously necessary, and in a number of diseases involving defects in liver specific enzymes.
Setting DNA tests in a proper perspective
DNA testing for a disease should only proceed after the individual family has received adequate genetic counselling and has achieved a clear understanding of the nature and meaning of the results that they may receive from the test. If the doctor does not feel confident to give this counselling, then the patient should be referred to a clinical genetics service. The technology of DNA testing is changing so rapidly that clinicians who are not geneticists should always confer with the local clinical genetics service before discussing DNA testing with a family to be sure the information they are intending to impart is up to date.
Sometimes couples seeking prenatal diagnosis* have not really thought through what they will do with the test result, especially if it is abnormal. This problem is even more striking in presymptomatic testing for genetic diseases of adult life, such as Huntington's disease. Often individuals do not realise that a test which can prove that they have not inherited the gene causing the disease may instead reveal that they are going to develop the disease. The implications of being a carrier of an X-linked condition are relatively easy to explain. It is much more difficult to convince people that the knowledge of carrier status for autosomal recessive conditions is of very little practical significance, except in the few very common recessive conditions, and is certainly not a reason for great anxiety.
Testing of children requires very careful consideration. In most situations, it is more appropriate to delay testing until they are old enough to make their own decisions. In many families, it becomes apparent that there are other family members who are at as much risk as the ones seeking advice. The feasibility and appropriateness of contacting these individuals to offer them counselling and testing will need to be assessed.
Another factor that clinicians must always remember is that considerable background investigation of the individual family may be necessary before it is possible to offer prenatal diagnostic tests. Couples who may wish to use this procedure should be referred well before they start a pregnancy. In diseases for which only linkage testing is available, it may sometimes turn out that the particular couple cannot be offered a test at all, because of their particular family structure or for other chance reasons.
At present, DNA tests have no item number in the Medicare Pathology Schedule. Direct funding of DNA diagnostic laboratories has been considered desirable to ensure close liaison with clinical geneticists who know the complexities and pitfalls of the tests and the importance of counselling families.
* Prenatal diagnosis has become the accepted term for tests performed during pregnancy to recognise fetal abnormalities.
Mutations causing diseases
Mutations can be (in order of decreasing frequency)
It is easy to develop a simple test for any of these mutations in other family members once a mutation has been discovered in the index case. Demonstration of the mutation in the index case is fairly straightforward if the mutation is a deletion, duplication or triplet repeat, but is often very difficult in single base pair mutations.
Types of DNA diagnostic tests
DNA testing falls into two broad groups direct detection of the causative mutation and indirect tests using linkage analysis (gene tracking).
Direct mutation detection
Whenever possible, direct mutation detection is performed. Results are unambiguous, individuals can be tested in isolation (without the need for samples from other family members) and prenatal diagnosis is always possible. This process starts by searching for gene mutations in an affected individual. In autosomal dominant or X-linked conditions, there will be just one mutation, but in autosomal recessive conditions, there may be two different mutations rather than two copies of the same mutation. The types of mutations found in different genetic diseases vary greatly and an appropriate strategy has to be chosen for each disease. In dominant and X-linked conditions, it may be justifiable to expend a considerable effort to find the mutation present in the family as this test can then be applied to all at risk family members. In autosomal recessive disorders, the test will only be useful within the nuclear family and so it is more usual to search for the frequent or easily detected mutations that are known and to turn to a linkage approach in families in which these mutations are not found.
Linkage (gene tracking)
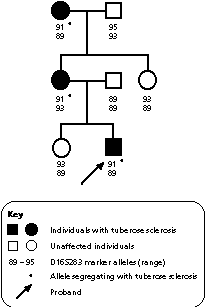
The logic here is to identify a testable marker in the DNA close to or within the gene concerned in causing the disease. Tests for this marker are used to track the mutant gene from parent to child. Fortunately, functionally insignificant variations in human DNA are very frequent, occurring about once in every 300 nucleotide bases. Therefore, it is generally possible to find an easily testable difference between the two chromosomes close to or within any gene of clinical importance. A number of types of DNA differences have been utilised. Currently, it is most usual to take advantage of runs of repeats of 2, 3 or 4 bases (microsatellite repeats) which are found in more than 100 000 locations in the human genome. The number of these repeats at each site is highly variable (polymorphic) and usually differs between the two chromosomes of each pair. Linkage is illustrated in Fig. 1.
|
Fig. 1 In this pedigree, the disorder (tuberose sclerosis) segregates with marker allele 91.
|
During meiotic division, genetic material can be exchanged between chromosomes. This exchange is also called recombination and can affect linkage studies if either the marker or the gene is exchanged without the other. The closer the marker is to the mutation within the gene, the less likely they are to be separated by recombination.
Methods for DNA testing
Collection and storage of DNA
Most DNA laboratories prefer blood to be collected in either heparin or EDTA tubes (this will be stated on the request form). For adults, 20 mL of blood is usually requested, but less is needed from children, because of their higher white blood count. When the polymerase chain reaction (PCR) is used, only very small amounts of blood may be necessary e.g. Guthrie card spots and single hair roots. Although lymphocytes are the most convenient source of DNA, almost any other tissue can be potentially used e.g. postmortem specimens and paraffin fixed slides. Once extracted, DNA is extremely stable and can be stored for decades with little deterioration. For this reason, it may be important to obtain and store samples from family members who may be needed in future direct mutation or linkage analysis studies.
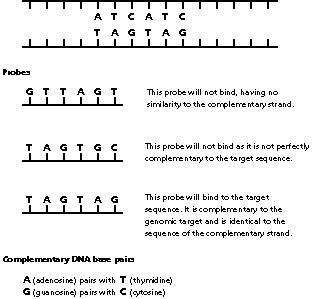
Genetic probes
Probes are nucleic acid bases arranged in a sequence which is complementary to a sequence in the genome (Fig. 2). They can be used to detect if a genomic sequence is present, deleted or altered. If this article had been written a few years ago, we would have detailed restriction fragment length polymorphisms (RFLPs). These are now rarely used, having been largely replaced by the far more useful repeat sequences.
|
Fig. 2 The two strands of genomic DNA are shown. The strand read by the cell is on top. It contains a sequence of importance to which the probe must bind. The complementary sequence is in the complementary strand underneath.
|
Probes can now be manufactured to detect any genomic locus or mutation if the DNA base sequence is known. Most probes detect only one locus in the human genome. A few deliberately detect multiple loci on different chromosomes, such as the Jeffrey's fingerprinting probes used to confirm paternity and in forensic studies.
|
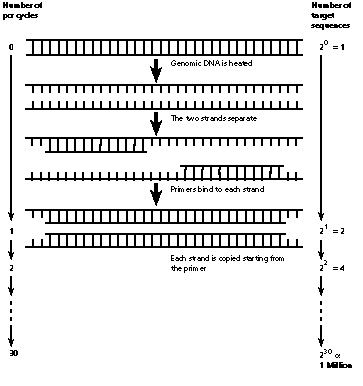
Fig. 3
|
Southern blotting and molecular hybridisation
The process of Southern blotting involves a DNA sample being cut at specific sites by enzymes known as restriction endonucleases. The fragments are size sorted by electrophoresis, covalently bound to a membrane and analysed with a radio labelled gene probe. Southern blotting is used for direct mutation detection, identifying deletions and duplications, and to produce linkage results.
Polymerase chain reaction (PCR)
The polymerase chain reaction is a method of amplifying a particular stretch of DNA, usually by a millionfold (Fig. 3). A pair of oligonucleotide primers, each usually about 20 bases long, is used. One binds to each end of the segment to be studied. A reaction with DNA polymerase is repeated 30-35 times, with a doubling of the DNA segment after each cycle. The polymerase chain reaction is the method of choice for DNA analysis as it is quicker than Southern blotting (hours versus 1-2 weeks), tiny quantities of DNA are used and the oligonucleotide primers can be synthesised in hours. The primers can be easily designed to identify specific mutations.
In situ hybridisation and fluorescent in situ hybridisation (FISH)
In situ hybridisation is a technique used to detect deletions and rearrangements of chromosomal DNA. A probe specific for the area of interest is applied to a cytogenetic chromosome preparation. In deletion detection, in situ hybridisation will show two signals in a normal individual, one on each locus. In an individual with a deletion, only one signal is seen and this is on the normal chromosome. There is no sequence for the probe to bind to on the chromosome with the deletion and therefore no signal. The number of conditions that are recognised to be caused by deletions is increasing and in situ hybridisation may well become the method of choice for their detection.
If a chromosomal rearrangement is being sought, then a library (collection) of probes for the area of interest is used.
Individual chromosome libraries each contain probes from multiple loci on the chromosome of interest and will 'paint' all of the chromosome. The presence and/or derivation of a chromosome rearrangement can be ascertained by observing which parts of the chromosomes react (are painted) with the probes. This is particularly important for the detection of translocations because of the risk of children being born with an imbalanced karyotype. This technique is being investigated to attempt prenatal diagnosis for trisomies 21, 18 and 13.
DNA results, reporting and interpretation
DNA laboratories typically describe the mutation detected and the methodology used, or linkage results and the probes used, and give an interpretation of the result. At present, most DNA tests are performed via a clinical genetics department and the family is counselled by a clinical geneticist. As the diseases which can be tested for and the number of tests which are performed have increased, and as direct mutation detection has become more widely available compared to linkage analysis, results are now often sent to the referring clinician who takes responsibility for counselling.
The detection of nonpaternity during DNA investigations may have significant ethical consequences and requires particularly sensitive counselling. In addition, the risk to the individual being tested may be reduced or be unable to be determined.
It is important for the clinician to understand the reliability of the test and its implications for the individual and their family. In common with other investigations, genetic tests can occasionally give complex or misleading results. A clinical genetics department should be involved if there are any difficulties.
Further reading
Weatherall DJ. The new genetics and clinical practice. 3rd ed. Oxford: Oxford University Press, 1991.
Emery AE, Mueller RF. Elements of medical genetics. 8th ed. Edinburgh: Churchill Livingstone, 1992.
Editor's note:
The next issue of Australian Prescriber will feature the clinical applications of DNA diagnostic tests.
The following statements are either true or false.
1. Deletions of part of a gene are the commonest type of mutation.
2. Gene probes cannot detect deletions.
Answers to self-test questions
1. False
2. False
Clinical Geneticist, The Murdoch Institute for Research into Birth Defects, Royal Children's Hospital, Melbourne
Professor and Director, The Murdoch Institute for Research into Birth Defects, Royal Children's Hospital, Melbourne