Experimental and Clinical Pharmacology
PCSK9 inhibitors – mechanisms of action
- Michael M Page, Gerald F Watts
- Aust Prescr 2016;39:164-7
- 1 October 2016
- DOI: 10.18773/austprescr.2016.060
PCSK9 is a proprotein convertase which is involved in the degradation of low-density lipoprotein (LDL) receptors in the liver.
Mutations in the PCSK9 gene cause familial hypercholesterolaemia in a subset of patients by reducing the number of LDL receptors on the surface of hepatocytes. This decreases their ability to clear LDL cholesterol from plasma.
Conversely, other PCSK9 mutations result in unusually low concentrations of plasma LDL cholesterol and a reduced risk of atherosclerotic disease.
Blocking the activity of PCSK9 with monoclonal antibodies reduces the degradation of LDL receptors and increases the clearance of LDL cholesterol.
An injection of PCSK9-specific antibody suppresses LDL-cholesterol concentrations for several weeks.
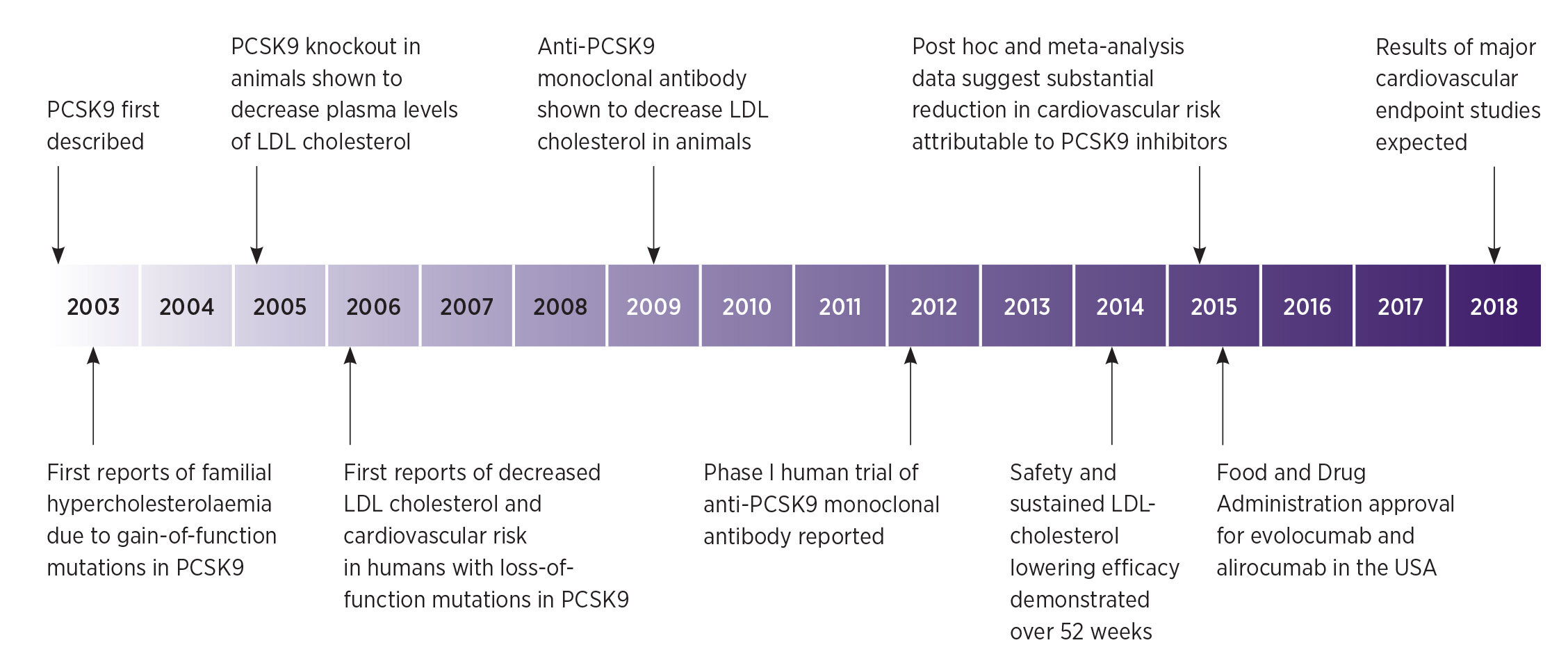
The 1985 Nobel Prize for Physiology or Medicine was awarded to Michael Brown and Joseph Goldstein for their research into the link between cholesterol metabolism and coronary artery disease.1 This research increased our understanding of the pathophysiology of disorders such as familial hypercholesterolaemia, and paved the way for important therapies like statins (HMG-CoA reductase inhibitors). They found that the low-density lipoprotein (LDL) receptor, expressed primarily in the liver, was responsible for clearing LDL particles from plasma.
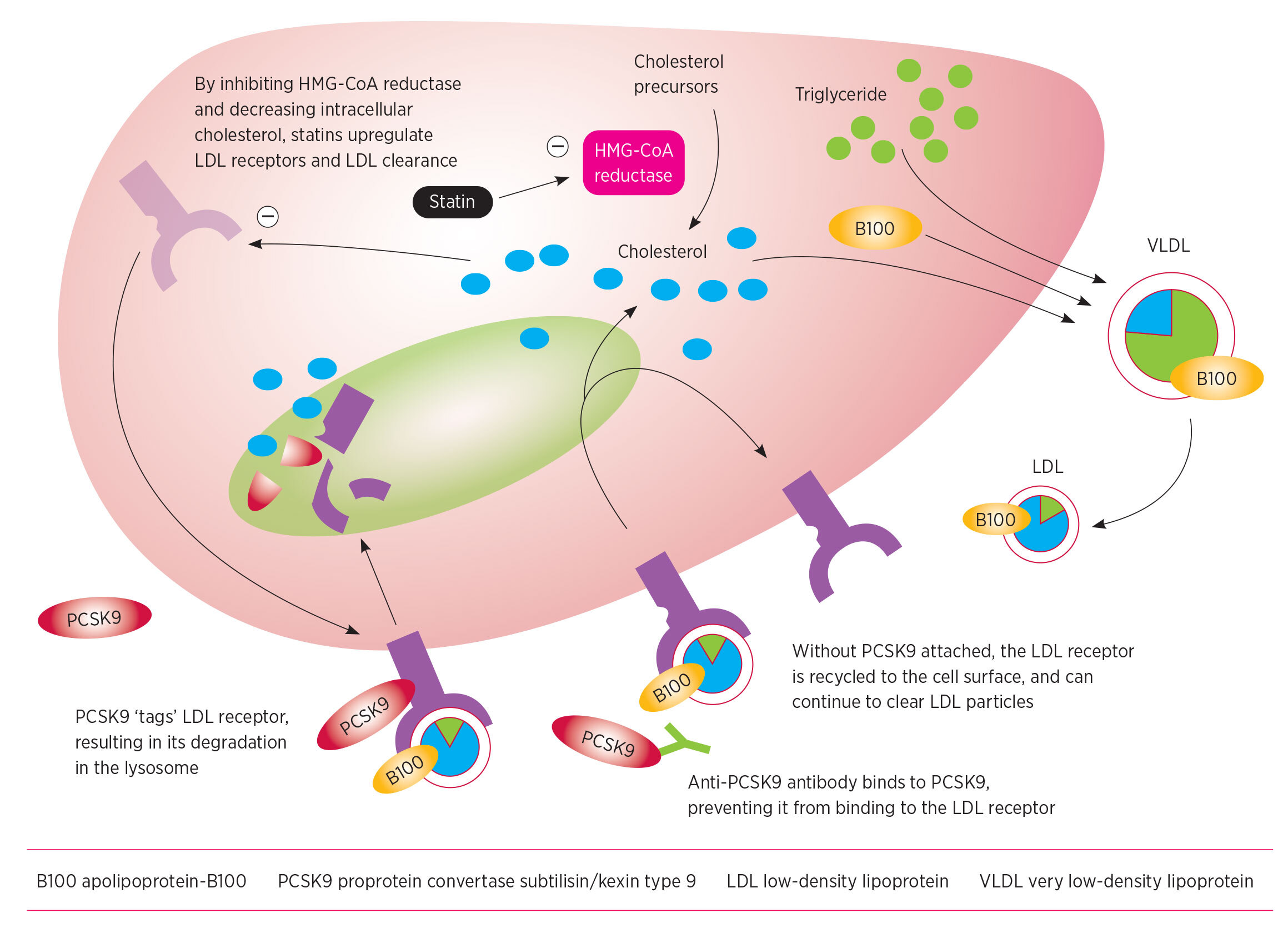
Statins decrease the intracellular concentration of cholesterol in the liver. This increases the expression of LDL receptors and more LDL cholesterol is removed from the circulation (Fig. 1).
VLDL is secreted by the liver and converted to LDL, which delivers cholesterol to peripheral tissues and is atherogenic. LDL particles are taken up via LDL receptors, primarily on hepatocytes, and degraded. The production of LDL receptors is decreased by intracellular cholesterol, so lowering intracellular cholesterol with statins results in increased LDL receptors and LDL uptake. LDL-receptor degradation is enhanced by PCSK9, so inhibiting PCSK9 with antibodies increases LDL-receptor recycling and LDL uptake.
Proprotein convertases are a family of enzymes involved in converting precursors of secretory proteins, such as hormones, enzymes and receptors, into bioactive molecules at their intended target tissue. These enzymes are part of regulatory pathways that help the body to maintain homeostasis.
LDL low-density lipoprotein
Most cases of familial hypercholesterolaemia are due to mutations resulting in defective LDL receptors, with others caused by defects in the ligand for the LDL receptor, apolipoprotein-B100. Inherited ‘gain-of-function’ mutations in the PCSK9 gene have recently also been found to cause familial hypercholesterolaemia. This is characterised by very high plasma concentrations of LDL cholesterol and associated with premature atherosclerotic cardiovascular disease.3 Conversely, ‘loss-of-function’ PCSK9 mutations result in unusually low concentrations of plasma LDL cholesterol. People with these mutations have a markedly reduced lifetime risk of atherosclerotic cardiovascular disease.4
PCSK9 circulates in three main forms:
Some PCSK9 mutations result in changes to the self‑association or furin-mediated cleavage of PCSK9. These mutations lead to gain or loss of function.5
PCSK9 is cleared from the circulation by the LDL receptor. It is then cleaved inside hepatocytes (Fig. 1).
PCSK9 regulates the degradation of the LDL receptor in response to cholesterol concentrations within the cell (Fig. 1). PCSK9 binds to an extracellular part of the LDL receptor. Apolipoprotein-B100, the structural protein of LDL and ligand for the LDL receptor, binds to a different site on the LDL receptor.
After the LDL receptor is internalised into the hepatocyte, it is trafficked to a lysosome, where it can be either degraded or recycled back to the surface of the hepatocyte. PCSK9 prevents the LDL receptor from forming a closed conformation, making the receptor susceptible to enzymatic degradation.6 LDL receptors without PCSK9 bound to them are therefore more likely to be recycled to the cell surface.
Although animal models suggest that PCSK9 has other roles in cholesterol metabolism aside from the regulation of LDL-receptor recycling, this has not been borne out in the clinical setting in humans. In LDL-receptor knockout mice, LDLcholesterol concentrations are increased by PCSK9 administration or overexpression, despite the absence of LDL receptors as a clearance pathway.7,8 However, this finding is inconsistent with that in humans with LDL‑receptor-negative familial hypercholesterolaemia, in whom blocking the action of PCSK9 does not decrease plasma concentrations of LDL cholesterol.9
Animal studies have suggested roles for PCSK9 in non-hepatic tissue. These include intestinal and adipocyte lipid metabolism, development of atherosclerotic plaques and inflammation, apoptotic cell death, and regulation of blood pressure and glycaemia.5 Clinically significant roles of PCSK9 other than in cholesterol metabolism have not been identified or emerged in the form of unexpected adverse effects despite thousands of patients being treated with anti-PCSK9 antibodies.
Inhibiting PCSK9 means that more LDL receptors will be recycled to the surface of the cell. This should increase the clearance of LDL cholesterol from the circulation.
Various approaches to the pharmacological inhibition of PCSK9 have been investigated. Molecules that prevent the formation of PCSK9 include antisense oligonucleotides and small interfering RNAs. Molecules that bind to mature PCSK9, preventing it from interacting with LDL receptors, include the small adnectin polypeptides and monoclonal antibodies.5
The anti-PCSK9 monoclonal antibodies are of most therapeutic interest and are currently in phase III trials. Details of their binding sites have not been fully disclosed, but earlier monoclonal antibodies are known to bind at or near PCSK9's binding site for the LDL receptor. In pre-clinical studies this sterically inhibits the interaction of PCSK9 with the LDL receptor.10 Blocking the binding of PCSK9 to the LDL receptor reduces the degradation of the receptor. This markedly increases the clearance of LDL and substantially lowers plasma LDL cholesterol, as well as apolipoprotein-B100 (Fig. 1).
Consistent with the long plasma half-life of monoclonal antibodies, a single dose of an anti-PCSK9 antibody can suppress the plasma LDLcholesterol concentration for several weeks. Repeated injections cause a sustained reduction of about 50–70% from baseline, as monotherapy or when added to a
statin.11-13
PCSK9 inhibition also decreases the plasma concentrations of lipoprotein(a) (Lp(a)) by around 20–30%.11,13 This particle is similar in size and cholesterol content to LDL. Unlike LDL, its apolipoprotein-B100 moiety is covalently linked to apolipoprotein(a), a potentially prothrombotic apolipoprotein with sequence similarity to plasminogen. Genetic and epidemiological studies suggest a causal association between Lp(a) and the risk of atherosclerotic cardiovascular disease.14
How PCSK9 inhibitors lower Lp(a) concentrations is unknown and warrants further research. Lp(a) is not currently understood to be cleared by the LDL receptor, and statins, which upregulate the LDL receptor, do not substantially lower plasma concentrations of Lp(a). However, recent in vitro studies have suggested that the LDL receptor may indeed have a role in Lp(a) clearance.15
The story of PCSK9 since its discovery just over a decade ago is an important case study in translating research into practice. PCSK9-inhibiting therapies have efficacy in lowering LDL cholesterol which could decrease the risk of atherosclerotic cardiovascular disease, particularly in high-risk patients. They could reduce the need for radical therapies such as lipoprotein apheresis in patients with severe heterozygous familial hypercholesterolaemia, and homozygous familial hypercholesterolaemia with residual LDL-receptor function.
Gerald Watts has received honoraria for advisory boards and research grants from Sanofi and Amgen.
Chemical pathology registrar, PathWest Laboratory Medicine, Fiona Stanley Hospital, Perth
Winthrop professor of cardiometabolic medicine, School of Medicine and Pharmacology, University of Western Australia, Royal Perth Hospital
