
What is in this leaflet
This leaflet answers some common questions about FONAT.
It is particularly important that you read the sections "When to take it" and "How to take it" before you take this medicine.
This leaflet does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have benefits and risks. Your doctor has weighed the risks of you taking FONAT against the benefits expected for you.
If you have any concerns about taking this medicine, talk to your doctor or pharmacist.
Keep this leaflet with your medicine. You may need to read it again.
What FONAT is used for
FONAT is used to treat osteoporosis.
This condition is caused by changes in the way bone is normally maintained.
How does FONAT work?
FONAT belongs to a group of non-hormonal medicines called bisphosphonates.
FONAT works by slowing down the process of old bone being removed, which allows the bone-forming cells time to rebuild normal bone. FONAT not only helps prevent the loss of bone but actually helps to rebuild bone and makes bone less likely to fracture. Thus, FONAT prevents or reverses the progression of osteoporosis.
FONAT starts working on the bone cells immediately, but measurable effects on bone mass may not be seen for several months or more.
Understanding bone
Bone is living, growing tissue. Throughout life, our bodies are breaking down old bone and rebuilding new bone in a continuous cycle. Until our late 20s, while bones are still developing, we gain bone by building more than we lose. From then until about age 35 the process is usually in balance, so that the amount of bone lost is about equal to the amount that is replaced. This balanced process keeps your skeleton healthy and strong. After about age 35 this balance is disturbed, with bone loss occurring at a slightly faster rate than it can be replaced. In women, after menopause, hormonal changes cause bone loss at an even faster rate. When bone loss is excessive, bones can become thinner and weaker, and therefore are more likely to break.
Osteoporosis
"Osteo" means bone, and "porosis" means something that has holes in it, like a sponge. Therefore, osteoporosis is a disease which causes bones to become more porous, gradually making them weaker, more brittle and likely to break.
Osteoporosis is common in postmenopausal women. The menopause occurs when the ovaries virtually stop producing the female hormone, oestrogen, or are removed (which may occur, for example, at the time of a hysterectomy). At this time, bone is removed faster than it is formed, so bone loss occurs and bones become weaker. The earlier a woman reaches the menopause, the greater the risk of osteoporosis.
Osteoporosis also occurs in men but is less common than in women.
Osteoporosis can also occur in people receiving corticosteroid medicines. If taken in high doses or for a long period of time, corticosteroid medicines can cause bone to be removed faster than it is formed. This causes loss of bone and therefore, bones become weaker and are more likely to break.
Maintaining bone mass and preventing further bone loss are important to keep your skeleton healthy.
Early on, osteoporosis usually has no symptoms. However, if left untreated it can result in broken bones, also called fractures.
Although fractures usually cause pain, fractures of the bones of the spine may go unnoticed until they cause height loss. Fractures may occur during normal, everyday activity, such as lifting, or from minor injury that would not ordinarily fracture normal bone.
Fractures usually occur at the hip, spine, or wrist and can lead not only to pain, but also to considerable deformity and disability, such as stooped posture from curvature of the spine, and loss of mobility.
Ask your doctor if you have any questions about why FONAT has been prescribed for you. Your doctor may have prescribed FONAT for another reason.
FONAT is available only with a doctor's prescription.
FONAT is not addictive.
Before you take FONAT
You should know that in some people, FONAT can irritate or burn the food pipe (also called oesophagus). The chances of this happening should be reduced when you follow the instructions for 'How to take FONAT' on the next page.
When you must not take it
Do not take FONAT if:
- You have an allergy to alendronate or any of the ingredients listed at the end of this leaflet
- you have certain disorders of the food pipe (oesophagus) including those that cause difficulty in swallowing
- you are unable to stand or sit upright for at least 30 minutes
- your doctor has told you that you currently have low blood calcium
- your dentist advises you to consult your doctor first.
Do not take FONAT if you are pregnant or breast-feeding. FONAT has not been studied in pregnant or breast-feeding women.
Do not give FONAT to a child. Safety and effectiveness of this medicine in children have not been established.
Do not take FONAT after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking FONAT, talk to your doctor.
Before you start to take it
Tell your doctor if:
- you are pregnant or you plan to become pregnant or breast-feeding.
- you have any medical conditions, especially the following:
- kidney disease
- swallowing or digestive problems, such as ulcers - you have any allergies to any other medicines, foods, preservatives or dyes.
- you have dental or jaw-bone problems or are planning to have a course of dental surgery
- you currently smoke or have been a smoker in the past.
If you have not told your doctor about any of the above, tell him/her before you start taking FONAT.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from a pharmacy, supermarket or health food shop.
Some medicines are likely to interfere with the absorption of FONAT if taken at the same time. These include:
- antacids, medicines used to treat indigestion
- calcium supplements
- vitamins
These medicines may be affected by FONAT or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Therefore, take FONAT at least 30 minutes before taking any of these or other medicines to make sure there is no problem with absorption.
Check with your doctor or pharmacist if you are not sure whether you are taking any of these medicines.
You can take aspirin while you are being treated with FONAT. However, both aspirin and FONAT may increase the chance of stomach upsets.
Your doctor or pharmacist has more information on medicines to be careful with or avoid while taking FONAT.
How to take FONAT
Follow all directions given to you by your doctor and pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box/bottle, ask your doctor or pharmacist for help.
How much to take
Take FONAT only when prescribed by your doctor.
The usual dose of FONAT is one 70 mg tablet once a week.
How to take it
Take FONAT after getting up for the day. Do not take it at bedtime.
Swallow one tablet whole with a full glass of plain water only. Do not take any food, medicines or drinks other than plain tap water with your FONAT. It is important to take FONAT with plain water only, not mineral water.
Food, other drugs, mineral water and other drinks, including fruit juices, coffee and tea, will reduce the effect of FONAT by interfering with the absorption into the body.
Stay upright for at least 30 minutes after swallowing FONAT and do not take any food, medicines or drinks other than plain tap water during this time.
Do not lie down immediately after swallowing it. It is important to stay upright (sitting, standing or walking around) for at least 30 minutes after swallowing your tablet.
It is also very important to stay upright until after you have eaten your first food of the day.
These actions will help make sure your tablet reaches your stomach quickly and help reduce the potential for irritation to your food pipe (oesophagus).
FONAT is effective only if taken when your stomach is empty. Food, drinks other than plain water, and other medicines will lessen the effect of FONAT by interfering with its absorption into the body.
Do not chew or suck on a tablet of FONAT. Mouth ulcers may occur if the tablet is chewed or dissolved in the mouth.
When to take it
Choose the day of the week that best fits your schedule. Every week, take one table of FONAT on your chosen day.
How long to take it for
It is important that you continue taking FONAT for as long as your doctor prescribes.
FONAT can only treat your osteoporosis, by helping prevent further loss of bone and continuing to rebuild bone, if you take it every week.
If you forget to take it
Skip the dose you missed and take your next dose the following morning.
If you take the forgotten tablet after you have eaten or had a drink, FONAT will not work as well as it should.
Therefore, it is better to skip the dose that you missed.
If you are not sure about what to do, talk to your doctor or pharmacist.
Do not take a double dose to make up for the dose that you missed.
If you have trouble remembering to take your FONAT, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much FONAT. Do this even if there are no signs of discomfort or poisoning.
If you take too many tablets at one time, drink a full glass of milk. Do not induce vomiting. Do not lie down.
While you are taking FONAT
Things you must do
If you develop difficulty or pain upon swallowing, chest pain, or new or worsening heartburn, stop taking FONAT and call your doctor.
If you become pregnant while taking FONAT, stop taking the tablets and tell your doctor.
Tell your doctor and pharmacist that you are taking FONAT, if you are about to be started on any new medicine.
Tell your dentist that you are taking FONAT, if you develop a toothache or require a dental procedure.
Tell your doctor, if you develop new or unusual pain in your leg. Rarely, patients have experienced fracture in a specific part of the thigh bone.
Make sure you have an adequate intake of calcium in your diet. Your doctor, dietician or pharmacist can tell you what foods you should eat.
Visit your doctor regularly so they can check on your progress.
Things you must not do
Do not give FONAT to anyone else, even if they have the same condition as you.
Things to be careful of
There have been side effects reported with alendronate that may affect your ability to drive or operate machinery. Individual responses to alendronate may vary. (See Side Effects).
Things that would be helpful for your osteoporosis
Some self help measures suggested below may help your osteoporosis.
Talk to your doctor or pharmacist about these measures and for more information.
- Exercise - can be helpful in building and maintaining strong bones. Regular exercise such as a brisk walk is a good idea. Talk to your doctor before you begin any exercise program.
- Diet - eat a balanced diet. You may need to increase the amount of calcium in your diet by eating calcium-rich foods or taking a calcium supplement. Your doctor will advise you.
- Smoking - appears to increase the rate at which you lose bone and, therefore, may increase your risk of fracture. Your doctor may ask you to stop smoking or at least cut down.
- Alcohol - your doctor may advise you to cut down the amount of alcohol you drink. If you drink excessively on a regular basis, you may increase your risk of developing osteoporosis.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking FONAT.
FONAT helps most people with osteoporosis, but it may have unwanted side effects in a few people.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
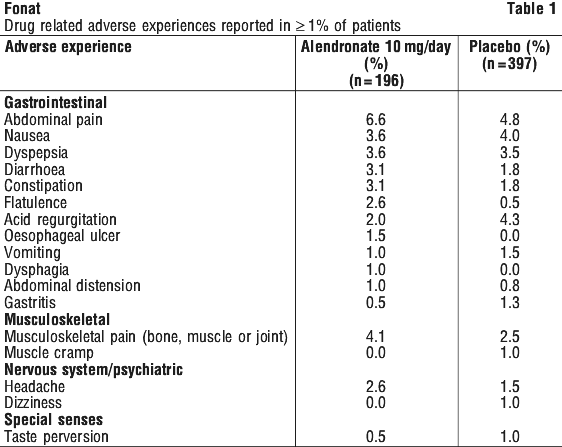
Tell your doctor if you notice any of the following and they worry you:
- stomach pain, gas in the stomach or bowel, wind
- an uncomfortable feeling in the stomach or belching after eating, also called dyspepsia, or heartburn
- feeling sick (nausea), vomiting
- constipation, diarrhoea
- headache
- unusual tiredness or weakness
- aching muscles, joints and/or bones, which rarely can be severe.
- flu-like symptoms typically at the start of treatment, such as aching muscles, generally feeling unwell and rarely fever.
- swelling of joints
- dizziness or spinning sensation
- swelling of hands, ankles or feet
- hair loss
- changes sense of taste
Most of these are the more common side effects of FONAT. For the most part, these have been mild.
Tell your doctor immediately if you notice any of the following:
- skin rash or redness of the skin, sometimes made worse by sunlight, itchiness
- mouth ulcers
- blurred vision, pain or redness in the eye
- symptoms of low blood calcium levels including muscle cramps or spasms or tingling sensation in the fingers or around the mouth
- new or unusual pain in your hip or thigh.
These side effects are rare, and very rarely, may be serious.
Tell your dentist and doctor immediately if you notice any of the following:
- Jaw-bone or dental problems (including toothache). Jaw-bone problems may include infection, and delayed healing after a tooth extraction or other work that involves drilling into the jaw-bone.
These side effects are rare and may be serious.
If any of the following happen, stop taking FONAT and tell your doctor immediately:
- difficulty or pain upon swallowing
- chest pain
- new or worsening heartburn
These side effects may be due to irritation or ulceration of the food pipe. They may worsen if you continue taking the tablets. Rarely, these side effects may be serious.
If any of the following happen, stop taking FONAT and tell your doctor immediately or go to accident and emergency at your nearest hospital:
- swelling of the face, lips, mouth, throat or tongue which may cause difficulty in breathing or swallowing
- pinkish, itchy swellings on the skin, also called hives or nettlerash
- severe skin reactions
- black tar-like and/or bloody stools
These may be serious side effects.
You may need urgent medical attention. These side effects are rare. If you have the swelling described above, you may be having a serious allergic reaction to FONAT.
Rarely, stomach or duodenal ulcers (some severe) have occurred, but it is not known whether these were caused by FONAT.
Other side effects not listed above may also occur in some patients.
Tell your doctor if you notice any other effects. Do not be alarmed by this list of possible side effects. You may not experience any of them.
After taking FONAT
Storage
Keep your tablets in the blister pack until it is time to take them. If you take the tablets out of the blister pack they may not keep well.
Keep FONAT in a cool dry place where the temperature stays below 25°C.
Do not freeze the product.
Do not store it or any other medicine in the bathroom or near a sink.
Do not leave it in the car or on window sills. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
FONAT comes as a white bi-convex tablet, debossed "AD70" on one side and "G" on the reverse.
FONAT is available in a blister pack containing 4 tablets.
Ingredients
Active ingredient:
- 91.37 mg alendronate sodium trihydrate equivalent to 70 mg alendronic acid per tablet
Inactive ingredients:
- lactose monohydrate
- microcrystalline cellulose
- povidone
- croscarmellose sodium
- magnesium stearate
FONAT contains trace quantities of sulfites and sugars as lactose.
Supplier
FONAT is supplied in Australia by:
Alphapharm Pty Ltd trading as Viatris
Level 1, 30 The Bond
30-34 Hickson Road
Millers Point NSW 2000
www.viatris.com.au
Phone: 1800 274 276
Australian registration numbers:
FONAT 70 mg blister pack - AUST R 134702
This leaflet was prepared in April 2024.
FONAT_cmi\Apr24/00
Published by MIMS June 2024
 Rarely, rash and erythema have occurred.
Rarely, rash and erythema have occurred.

 These increases were highly significant relative both to baseline and placebo at each measurement site in each study. Increases in BMD were evident as early as three months and continued throughout the entire three years of treatment. See Figure 2 for lumbar spine results. In the two year extension of these studies, treatment with alendronate 10 mg/day resulted in continued increases in BMD at the lumbar spine and trochanter (absolute additional increases between years 3 and 5: lumbar spine 0.94%; trochanter 0.88%). BMD at the femoral neck, forearm and total body were maintained. Thus, alendronate appears to reverse the progression of osteoporosis as assessed by increased bone mineral density. Alendronate was similarly effective regardless of age, race, baseline rate of bone turnover, renal function and use of concomitant medications.
These increases were highly significant relative both to baseline and placebo at each measurement site in each study. Increases in BMD were evident as early as three months and continued throughout the entire three years of treatment. See Figure 2 for lumbar spine results. In the two year extension of these studies, treatment with alendronate 10 mg/day resulted in continued increases in BMD at the lumbar spine and trochanter (absolute additional increases between years 3 and 5: lumbar spine 0.94%; trochanter 0.88%). BMD at the femoral neck, forearm and total body were maintained. Thus, alendronate appears to reverse the progression of osteoporosis as assessed by increased bone mineral density. Alendronate was similarly effective regardless of age, race, baseline rate of bone turnover, renal function and use of concomitant medications. In patients with postmenopausal osteoporosis treated with alendronate 10 mg/day for one or two years the effects of treatment withdrawal were assessed. Following discontinuation, there were no further increases in bone mass and the rates of bone loss were similar to those in the placebo groups. These data indicate that continuous treatment with alendronate is required to produce progressive increases in bone mass.
In patients with postmenopausal osteoporosis treated with alendronate 10 mg/day for one or two years the effects of treatment withdrawal were assessed. Following discontinuation, there were no further increases in bone mass and the rates of bone loss were similar to those in the placebo groups. These data indicate that continuous treatment with alendronate is required to produce progressive increases in bone mass. Furthermore, in this population of patients with baseline vertebral fracture, treatment with alendronate significantly reduced the incidence of hospitalisations resulting from any cause (25.0% versus 30.7%, a 20% relative risk reduction). This difference appears to be related, at least in part, to the reduction in fracture incidence.
Furthermore, in this population of patients with baseline vertebral fracture, treatment with alendronate significantly reduced the incidence of hospitalisations resulting from any cause (25.0% versus 30.7%, a 20% relative risk reduction). This difference appears to be related, at least in part, to the reduction in fracture incidence.
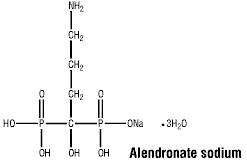 Chemical Name: (4-amino-1-hydroxybutylidene) bisphosphonic acid monosodium salt trihydrate.
Chemical Name: (4-amino-1-hydroxybutylidene) bisphosphonic acid monosodium salt trihydrate.