

What is in this leaflet
This leaflet answers some common questions about TARKA.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking TARKA against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What TARKA is used for
Tarka is used to treat hypertension (high blood pressure).
There are usually no symptoms of hypertension. The only way of knowing that you have hypertension is to have your blood pressure checked on a regular basis. If high blood pressure is not treated it can lead to serious health problems. You may feel fine and have no symptoms, but eventually hypertension can lead to serious health problems, including stroke, heart disease and kidney failure.
Tarka contains two different types of medicines; verapamil hydrochloride in a slow release formulation, and trandolapril in an immediate release formulation.
- Verapamil belongs to a group of medicines called calcium channel blockers.
Calcium channel blockers work by opening up blood vessels, which lets more blood and oxygen reach the heart and at the same time lowers high blood pressure. - Trandolapril belongs to a group of medicines called Angiotensin Converting Enzyme (ACE) Inhibitors.
ACE inhibitors work by relaxing your blood vessels, making it easier for blood to move around your body. This helps lower blood pressure and increase the supply of blood and oxygen to your heart.
Ask your doctor if you have any questions about why Tarka has been prescribed for you. Your doctor may have prescribed it for another reason.
There is no evidence that Tarka is addictive.
This medicine is available only with a doctor's prescription.
Tarka should not be given to children under the age of 18, as there have been no studies of its effects in children.
Before you take TARKA
When you must not take it
Do not take TARKA if you have an allergy to:
- any medicine containing trandolapril (or any other ACE inhibitor)
- any medicine containing verapamil.
- any of the ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction include skin rash, itching, shortness of breath or swelling of the face, lips or tongue, which may cause difficulty in swallowing or breathing.
Do not take Tarka if you have experienced symptoms such as wheezing, swelling of the face, lips, mouth, tongue, throat, hands or feet, intense itching or severe skin rashes with previous ACE inhibitor treatment or if you or a member of your family have had these symptoms either spontaneously, or in response to another medicine in the past (a condition called angioedema). Taking Tarka could cause this problem to happen again.
Do not take this medicine if you are pregnant or planning to become pregnant. It may affect your developing baby if you take it during pregnancy.
Do not breast-feed if you are taking this medicine. The active ingredients in TARKA may pass into breast milk and there is a possibility that your baby may be affected.
Do not give this Tarka to a child under the age of 18 years. Tarka's safety and effectiveness in children younger than 18 years have not been established.
Do not take TARKA if you:
- have severe kidney or liver problems
- have certain heart conditions (such as heart failure, a very slow heart rate, heart conduction problems, some irregular heartbeats, or disease of the heart muscle)
- have low blood pressure, (hypotension)
- are currently being treated with intravenous β-adrenoreceptor antagonists (with the exception of being treated in ICU)
- undergo treatments where your blood is treated outside your body (extracorporeal treatments), such as haemodialysis with certain membranes, or LDL-apheresis (removal of LDL from your blood)
- have a history of angioedema (if you or a member of your family have had wheezing, swelling of the face, lips, mouth, tongue, throat, hands or feet, intense itching or severe skin rashes, either spontaneously, with previous ACE inhibitor treatment or in response to another medicine in the past)
- are taking any of the following medications, or medications containing these ingredients:
- aliskiren (under certain conditions)
- ivabradine
- Direct Oral Anticoagulants (DOACs) which thin the blood such as dabigatran (under certain conditions)
- neutral endopeptidase (NEP) inhibitors such as sacubitril or racecadotril.
- sacubitril in combination with valsartan
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- any other heart problem, including aortic stenosis (narrowing of your aortic valve or aorta - the main artery leaving your heart)
- blood vessel (circulatory) disease or a stroke
- liver problems
- kidney problems, including narrowing of the arteries to your kidneys (called renal artery stenosis) or need dialysis
- high levels of potassium in your blood
- recent vomiting or diarrhoea or if you are dehydrated
- muscle conditions such as Duchenne muscular dystrophy, myasthenia gravis, Lambert-Eaton syndrome
- connective tissue disease
- dizzy spells
- diabetes
If you are of African origin, you may have a higher risk of angioedema.
In black patients, ACE inhibitors are less effective in lowering blood pressure than in white patients.
Tell your doctor if you are taking a diuretic ('fluid' tablets), potassium supplements, on a low-salt diet, or use potassium-salt substitutes.
Tell your doctor if you have or are about to have:
- surgery or general anaesthetic
- desensitisation treatment for an allergy e.g. to insect stings
- regular dialysis, blood filtration or similar procedures
- LDL removed from your blood (LDL-apheresis)
Tell your doctor or pharmacist if you are pregnant, intend to become pregnant or are breastfeeding. Your doctor can discuss with you the risks and benefits involved.
Tarka contains sugars (as lactose). Tell your doctor if you have an intolerance to some sugars.
If you have not told your doctor about any of the above, tell him/her before you start taking TARKA.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Several medicines that may cause unwanted reactions if used with Tarka are listed below. Please tell your doctor if you are taking any of the following:
Medicines used to treat heart problems or high blood pressure:
- Beta-blockers e.g. atenolol, propranolol, metoprolol, etc
- Diuretics (also called fluid tablets)
- Angiotensin II receptor blockers (also called ARBs, A2RAs, or sartans)
- Aliskiren
- Ivabradine
- Digoxin
- neutral endopeptidase (NEP) inhibitors such as sacubitril or racecadotril
- Any other medicines used to control an irregular heartbeat e.g. quinidine, flecainide, amiodarone, disopyramide, procainamide
- Any other medicines used to control high blood pressure
Medicines used to treat or prevent blood clots (sometimes referred to as "blood thinners")
- Direct Oral Anticoagulants (DOACs) which thin the blood such as dabigatran
- aspirin, heparin
Medicines used to lower cholesterol
- Statins such as atorvastatin, simvastatin or lovastatin
Medicines used to treat or prevent gout
- Colchicine, sulfinpyrazone, allopurinol
Medicines used to lower blood glucose:
- Anti-diabetic medicines such as insulin and any oral hypoglycaemic medicines, including metformin, glibenclamide or vildagliptin
Medicines used to treat psychological problems
- Any medicines used to treat depression, anxiety, or psychosis; such as imipramine, buspirone, midazolam, lithium or tricyclic antidepressants.
Medicines used to treat epilepsy or seizures:
- Phenytoin, carbamazepine, phenobarbital (phenobarbitone)
Medicines used to treat or prevent organ transplant rejection (immunosuppressants):
- Such as ciclosporin, everolimus, sirolimus, tacrolimus and temsirolimus
Medicines used to treat infections or tuberculosis
- Such as erythromycin, clarithromycin, telithromycin or rifampicin
- Co-trimoxazole (trimethoprim and sulfamethoxazole)
Medicines used in the treatment of Human Immunodeficiency Virus (HIV):
- Such as ritonavir
Medicines used in surgical procedures such as:
- General anaesthetics used for inducing sleep
- Muscle relaxants, including dantrolene
Medicines used to treat pain and inflammation (such as arthritis) or fever:
- Non-steroidal anti-inflammatory drugs (NSAIDs) such as aspirin, ibuprofen, diclofenac, meloxicam, indometacin, celecoxib
- Injectable gold
Other medicines that may react with Tarka:
- Potassium supplements, or large amounts of salt
- Potassium - containing salt substitutes in your food (check the label)
- Sympathomimetics - these may be found in some decongestants, cough / cold remedies and asthma medicines
- Theophylline, a medicine used to treat asthma
- Doxorubicin and cytostatic medicines, used to treat certain cancers
- Cimetidine and other antacids, medicines commonly used to treat stomach ulcers and reflux
- Neuromuscular blocking agents such as vecuronium
Avoid grapefruit juice, as this may increase the blood levels of verapamil.
This is not a complete list of medicines which may interfere with Tarka.
Your doctor or pharmacist has more information on medicines to be careful with or to avoid while taking Tarka. You may need to use different amounts of your medicine, or take different medicines.
How to take TARKA
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet. Your doctor will tell you how many tablets you will need to take each day and when to take them. This depends on your condition, and whether or not you are taking any other medicines.
If you do not understand the instructions on the box/bottle, ask your doctor or pharmacist for help.
How much to take
The usual dosage for Tarka is one tablet daily.
The maximum dose of Tarka is one 4/240 mg tablet once daily.
How to take it
Swallow the tablets whole with a full glass of water.
Do not crush or chew Tarka tablets.
When to take it
Take Tarka at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
Tarka should ideally be taken in the morning with or after food.
If you need to take an antacid, take it at least 2 hours before or 2 hours after your dose of Tarka.
How long to take it
Continue taking your medicine for as long as your doctor or pharmacist tells you.
Tarka helps control your blood pressure, but it does not cure it.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering when to take your medicine, ask your pharmacist for advice.
If you take too much (overdose)
Immediately telephone your doctor, or the Poisons Information Centre (Tel: 13 11 26), or go to Accident and Emergency at your nearest hospital, if you think you or anyone else may have taken too much Tarka.
Do this even if there are no signs of discomfort or poisoning.
You may need urgent medical attention.
Symptoms of an overdose may include: a slow heartbeat, palpitations, chest pain, anxiety, feeling faint, dizzy or light-headed, and collapsing.
While you are using TARKA
Things you must do
Have your blood pressure checked when your doctor says, to make sure Tarka is working for you.
If you feel light-headed or dizzy after taking your first dose of Tarka, or when your dose is increased, tell your doctor immediately.
Tell your doctor if you have excessive vomiting and/or diarrhoea while taking Tarka. The loss of water and salt from your body may cause your blood pressure to drop too much.
Drink plenty of water when you are using Tarka, especially if you sweat a lot (e.g. during hot weather or exercise). If you do not drink enough water while taking Tarka, you may feel faint or light-headed or sick. This is because your blood pressure is dropping suddenly. If you continue to feel unwell, tell your doctor.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Tarka.
Tell any other doctors, dentists and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon that you are taking this medicine.
If you are about to receive any dialysis, desensitisation treatment for an allergy e.g. to insect stings, or about to have LDL removed from your blood (LDL-apheresis), tell your doctor that you are taking this medicine.
If you become pregnant while you are taking this medicine, tell your doctor or pharmacist immediately.
Visit your doctor regularly so that they can check on your progress. Your doctor may ask you to have blood tests to check your liver from time to time to make sure the medicine is working and to prevent unwanted side effects.
Things you must not do
Do not take eat grapefruit or drink grapefruit juice whilst taking Tarka.
Do not use this medicine to treat any other complaints unless your doctor or pharmacist tells you to.
Do not give this medicine to anyone else, even if they have the same condition as you.
Do not stop taking Tarka, or lower the dosage, without checking with your doctor.
Things to be careful of
If you feel light-headed, dizzy or faint when getting out of bed or standing up, get up slowly. Standing up slowly, especially when you get up from bed or chairs, will help your body get used to the change in position and blood pressure. If this problem continues or gets worse, talk to your doctor.
Be careful driving or operating machinery until you know how Tarka affects you. Tarka may cause dizziness, light-headedness, or tiredness, in some people. Make sure you know how you react to Tarka; and if you have any of these symptoms, do not drive, operate machinery or do anything else that could be dangerous.
Avoid drinking alcohol while you are taking this medicine. You may experience greater blood pressure lowering effects than usual. You may experience worsening dizziness or light-headedness.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Tarka.
Like all other medicines, Tarka may have unwanted side effects in a few people. Sometimes they are serious, but most of the time they are not. You may need medical attention if you get some of the side-effects.
If you are over 65 years of age you may have an increased chance of getting side effects. Report any side effects to your doctor promptly.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- persistent dry cough
- taste disturbance
- constipation
- nausea, vomiting, diarrhoea or stomach upset/stomach pains
- dizziness, light-headedness
- headache
- unusual tiredness or weakness
- aching, tender, or weak muscles not caused by exercise
- flushing
The above list includes the more common side effects of your medicine.
Tell your doctor as soon as possible if you notice any of the following:
- slow, fast, or irregular heart beat
- shortness of breath (sometimes with tiredness, weakness and reduced ability to exercise), which may occur together with swelling of the feet and legs due to fluid build up
- fever, upper stomach pain, feeling generally unwell
- symptoms of sunburn which may occur more quickly than normal, severe blisters, skin rash, itching or flaking skin
- signs of frequent infections such as fever, severe chills, sore throat or mouth ulcers
The above list includes serious side effects that may require medical attention. Serious side effects are rare.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- swelling of the face, lips, mouth, tongue or throat which may cause difficulty in swallowing or breathing
- severe dizziness and confusion with visual disturbances and speech problems
- rapid, shallow breathing, cold clammy skin, a rapid, weak pulse, dizziness, weakness and fainting
- chest pain, fainting, collapse
The above list includes very serious side effects. You may need urgent medical attention. These side effects are rare.
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking TARKA.
Other side effects not listed above may occur in some people.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
After using TARKA
Storage
Keep your tablets in the pack until it is time to take them. If you take the tablets out of the blister pack, they may not keep well.
Keep your tablets in a cool dry place where the temperature stays below 25°C.
Do not store TARKA or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
Tarka comes in two types of tablets:
Tarka 2/180 - pink, oval, film coated tablets, marked with the numbers "182" on one side.
The 2/180 mg strength is available in boxes of 28 tablets.
Tarka 4/240 - red-brown, oval, film coated tablets, marked with the numbers "244" on one side.
The 4/240 mg strength is available in boxes of 28 tablets.
Ingredients
Tarka 2/180 contains 2 mg of trandolapril and 180 mg of slow release verapamil hydrochloride as the active ingredients.
Tarka 4/240 contains 4 mg of trandolapril and 240 mg of slow release verapamil hydrochloride as the active ingredients.
Tarka 2/180, and Tarka 4/240 tablets also contain:
- microcrystalline cellulose
- docusate sodium
- hyprolose
- hypromellose
- iron oxide black
- iron oxide red
- iron oxide yellow
- lactose monohydrate
- macrogol 400
- macrogol 6000
- magnesium stearate
- povidone
- colloidal anhydrous silica
- sodium alginate
- sodium stearylfumarate
- maize starch
- purified talc
- titanium dioxide
Tarka contains sugars (as lactose).
This medicine does not contain sucrose, gluten, tartrazine or any other azo dyes.
Manufacturer/Distributor/ Supplier
TARKA is supplied in Australia by:
Mylan Health Pty Ltd
Level 1, 30 The Bond
30-34 Hickson Road
Millers Point, NSW 2000
www.mylan.com.au
Phone: 1800 314 527
Australian registration numbers:
TARKA 2/180 mg tablets:
AUST R 104663
TARKA 4/240 mg tablets:
AUST R 104664
TARKA® = Registered trademark
This leaflet was prepared in July 2021.
Tarka_cmi\Jul21/00
Published by MIMS August 2021
 Additional significant adverse events seen with verapamil hydrochloride are listed by body system. See Table 2.
Additional significant adverse events seen with verapamil hydrochloride are listed by body system. See Table 2. Additional significant adverse events seen with trandolapril are listed by body system. See Table 3.
Additional significant adverse events seen with trandolapril are listed by body system. See Table 3.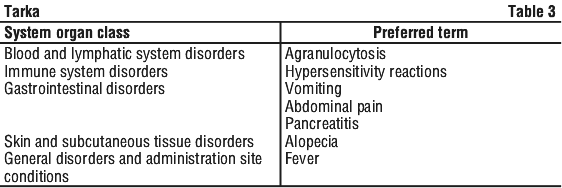 The following adverse events have been reported with ACE inhibitors as a class. See Table 4.
The following adverse events have been reported with ACE inhibitors as a class. See Table 4.


 Chemical name: (2S,3aR,7aS)-1-[(2S)-2-[[(1S)-1-(Ethoxycarbonyl)-3- phenylpropyl]amino]propanoyl]octahydro-1H-indole-2-carboxylic acid. C24H34N2O5, MW: 430.54.
Chemical name: (2S,3aR,7aS)-1-[(2S)-2-[[(1S)-1-(Ethoxycarbonyl)-3- phenylpropyl]amino]propanoyl]octahydro-1H-indole-2-carboxylic acid. C24H34N2O5, MW: 430.54. Chemical name: benzeneacetonitrile, α-[3-[{2-(3,4-dimethoxyphenyl)ethyl}methylamino] propyl]-3,4- dimethoxy-α -(1-methylethyl) monohydrochloride. C27H38N2O4.HCl, MW: 491.08.
Chemical name: benzeneacetonitrile, α-[3-[{2-(3,4-dimethoxyphenyl)ethyl}methylamino] propyl]-3,4- dimethoxy-α -(1-methylethyl) monohydrochloride. C27H38N2O4.HCl, MW: 491.08.