

What is in this leaflet
This leaflet answers some common questions about TECFIDERA. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
This leaflet was last updated on the date at the end of this leaflet.
Speak to your pharmacist or doctor to obtain the most up to date information on this medicine.
You can also download the most up to date leaflet from: www.biogen.com.au/products/tecfidera-CMI.pdf
All medicines have risks and benefits. Your doctor has weighed the risks of you taking TECFIDERA against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What TECFIDERA is used for
TECFIDERA is used to treat relapsing multiple sclerosis (MS).
TECFIDERA slows down the progression of physical disability in people with relapsing forms of MS and decreases the number of flare ups (relapses).
Some people feel better when they start to take TECFIDERA. However TECFIDERA cannot repair damage that has already been caused by MS. When you start TECFIDERA you might not notice an improvement, but TECFIDERA may still be working to help prevent your MS from becoming worse.
The cause of MS is not yet known, MS affects the brain and spinal cord. In MS, the body's immune system reacts against its own myelin (the 'insulation' surrounding nerve fibres). In relapsing forms of MS, people have 'exacerbations' from time to time (e.g. blurred vision, weakness in the legs or arms, or loss of control of bowel or bladder function). They are followed by periods of recovery. Recovery may be complete or incomplete. If it is incomplete there is 'progression of disability'.
TECFIDERA contains the active ingredient dimethyl fumarate. Dimethyl fumarate decreases the inflammation in your brain that is caused by MS and thereby reduces nerve damage.
TECFIDERA works by reducing inflammatory responses in cells and helps to protect the central nervous system cells against attack. Inflammation of the brain is an important part of the MS disease process.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
TECFIDERA has not been studied in patients with chronic progressive MS.
Safety and effectiveness in children younger than 18 years have not been established.
This medicine is not addictive.
This medicine is available only with a doctor's prescription.
Before you take TECFIDERA
When you must not take it
Do not take TECFIDERA if you have an allergy to:
- any medicine containing dimethyl fumarate
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
Do not take TECFIDERA if you are being treated with other medicines containing fumaric acid (creams or tablets/capsules).
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- liver problems
- kidney problems
- infection
- recently received a vaccination.
Tell your doctor at your earliest opportunity if you suspect you have any symptoms of shingles.
Tell your doctor if you are pregnant or intend to become pregnant. There is no information on the use of TECFIDERA during pregnancy. Your doctor will discuss the risks and benefits of taking it if you are pregnant.
Tell your doctor if you are breast-feeding or planning to breast-feed. It is not known whether TECFIDERA passes into breast milk. Your doctor will discuss the risks and benefits of taking it if you are breast-feeding.
If you have not told your doctor about any of the above, tell them before you start taking TECFIDERA.
Taking other medicines
Tell your doctor if you have previously taken or are currently taking medicines containing fumaric acid (creams or tablets/capsules).
You should not take TECFIDERA together with these medicines.
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and TECFIDERA may interfere with each other. These include:
- medicines which affect immune function including other medicines to treat MS such as fingolimod, natalizumab or mitoxantrone or some other commonly used cancer medicines
- medicines which affect the kidneys, including some antibiotics (used to treat infections), "water tablets" (diuretics), certain types of painkillers (such as ibuprofen and other similar anti-inflammatory medicines and medicines purchased without a doctor’s prescription) and medicines that contain lithium
- live vaccines.
These medicines may be affected by TECFIDERA or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take TECFIDERA
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
How much to take
The recommended starting dose of TECFIDERA is 120 mg taken twice daily. After 7 days the recommended dose is 240 mg twice daily.
How to take it
Swallow each capsule whole with a glass of water. Do not crush, divide or dissolve the capsule or its contents.
When to take it
Take one capsule twice a day.
Taking it at the same time each day (e.g. at morning during breakfast and at night during dinner) will help you remember when to take it.
TECFIDERA can be taken with or without food. For those patients who experience gastrointestinal side effects or flushing, taking TECFIDERA with food may help reduce these effects.
Your doctor may tell you to take TECFIDERA with aspirin or may temporarily reduce your dose.
Do not reduce your dose unless your doctor tells you to.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
This medicine helps to control your condition, but does not cure it. The positive effects of TECFIDERA may not be seen immediately. It is important to keep taking your medicine even if you feel well.
It is important not to interrupt treatment with TECFIDERA unless your doctor tells you to.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (in Australia telephone 13 11 26, in New Zealand telephone 0800 764 766) for advice, or go to Emergency at the nearest hospital, if you think that you or anyone else may have taken too much TECFIDERA. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are taking TECFIDERA
Things you must do
If you are about to have any blood or urine tests, tell your doctor that you are taking TECFIDERA.
Blood and urine test results may be affected by treatment with TECFIDERA.
Before you start TECFIDERA, your doctor will do a blood test to check the number of your white blood cells. Your doctor may also test these periodically during treatment.
Before you start TECFIDERA, your doctor will make sure you have results from a recent urine test to check your kidney function and may repeat the test periodically during treatment. TECFIDERA may cause proteins (such as albumin) to be detected in a urine test.
TECFIDERA may also cause increases in the level of liver enzymes that will show up in a blood test.
Take TECFIDERA exactly as your doctor has prescribed.
Tell your doctor if you are going to be vaccinated.
Tell your partner or caregiver about your treatment.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking TECFIDERA.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you become pregnant while taking this medicine, tell your doctor immediately.
Things you must not do
Do not take TECFIDERA to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor.
Things to be careful of
Tell your doctor straight away if you think you have an infection, have fever, or feel like you have the flu. TECFIDERA may decrease lymphocyte (white blood cell) counts. White blood cells fight infection. You may get infections more easily while you are taking TECFIDERA. Any infection that you already have may get worse. Infections could be serious and sometimes life-threatening. If you have a serious infection, your doctor may recommend that you stop taking TECFIDERA until you recover.
Tell your doctor straight away if you think you are experiencing symptoms similar to an MS relapse, new or worsening weakness on one side of the body; clumsiness; changes in vision, thinking, or memory; or confusion or personality changes lasting for more than several days. These could be signs of a rare and very serious brain infection called progressive multifocal leukoencephalopathy (PML). The symptoms of PML may be similar to an MS relapse.
Having low lymphocyte levels, particularly for a long period of time, can increase your risk of PML.
Keep all of your doctor's appointments so that your progress can be checked. Before you start TECFIDERA, your doctor will do a blood test to check the number of your white blood cells. Your doctor may also test these periodically during treatment.
Before you start TECFIDERA, your doctor will make sure you have results from a recent urine test to check your kidney function and may repeat the test periodically during treatment. TECFIDERA may cause proteins (such as albumin) to be detected in a urine test.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking TECFIDERA.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- reddening of the face or body feeling warm, hot, burning or itchy (flushing)
- loose stools (diarrhoea)
- feeling sick (nausea)
- stomach pain or stomach cramps
- inflammation of the lining of the intestines (gastroenteritis)
- being sick (vomiting)
- indigestion (dyspepsia)
- inflammation of the lining of the stomach (gastritis)
- gastrointestinal disorder
- burning sensation
- hot flush, feeling hot
- itchy skin (pruritus)
- rash
- pink or red blotches on the skin (erythema)
- runny nose (rhinorrhoea)
- hair thinning.
The above list includes the more common side effects of your medicine. If any of these persist or worsen, talk to your doctor as some of them may also be due to an infection or allergic reaction.
Tell your doctor as soon as possible if you notice the following:
- signs of infection (e.g., unexplained fever, severe diarrhoea).
The above list includes serious side effects that may require medical attention. Serious side effects are rare.
If any of the following happen, stop taking TECFIDERA and tell your doctor immediately or go to Emergency at your nearest hospital:
- swelling of your face, lips, tongue or other parts of the body
- shortness of breath, wheezing, difficulty breathing, chest pain or discomfort
- symptoms similar to an MS relapse, new or worsening weakness on one side of the body; clumsiness; changes in vision, thinking, or memory; or confusion or personality changes.
The above list includes very serious side effects. You may need urgent medical attention or hospitalisation. These side effects are very rare.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
After taking TECFIDERA
Storage
Keep your capsules in the pack until it is time to take them. If you take the capsules out of the pack they may not keep well.
Keep your capsules in a cool dry place away from light where the temperature stays below 30°C.
Do not store TECFIDERA or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
TECFIDERA capsules are available in two strengths: 120 mg and 240 mg.
The 120 mg capsules are green and white printed with 'BG-12, 120 mg' in black ink on the capsule body. Available in blister wallet cards containing 14 or 112 capsules packed in a box.
The 240 mg capsules are green printed with 'BG-12, 240 mg' in black ink on the capsule body. Available in blister wallet cards containing 14 or 56 capsules packed in a box.
Not all pack sizes may be available.
Ingredients
TECFIDERA contains dimethyl fumarate as the active ingredient.
Other ingredients:
- microcrystalline cellulose (E460)
- croscarmellose sodium (E468)
- purified talc (E553b)
- colloidal anhydrous silica (E551)
- magnesium stearate (E572)
- triethyl citrate (E1505)
- methylacrylate-methyl methacrylate copolymer
- methacrylic acid-ethyl acrylate copolymer
- simethicone
- sodium lauryl sulfate (E514)
- polysorbate 80 (E433)
- gelatin
- titanium dioxide (E171)
- brilliant blue FCF CI42090 (E133)
- iron oxide yellow CI77492 (E172)
- iron oxide black CI77499 (E172).
Further information
You can obtain more information from your doctor, pharmacist or the MS Society in your State, or by telephoning the MS Alliance on 1800 852 289 in Australia or 0800 852 289.
Sponsor
TECFIDERA is supplied in Australia by:
Biogen Australia Pty Ltd
Level 4, 2 Banfield Road
Macquarie Park NSW 2113
TECFIDERA is supplied in New Zealand by:
Biogen NZ Biopharma Limited
188 Quay Street
Auckland
® = Registered Trademark or
© Copyright
This leaflet was prepared in September 2021.
TECFIDERA 120 mg - AUST R 197118
TECFIDERA 240 mg - AUST R 197119
TECFIDERA® is a registered trademark of Biogen.
BIOGEN® is a registered trademark of Biogen MA Inc.
Published by MIMS November 2021
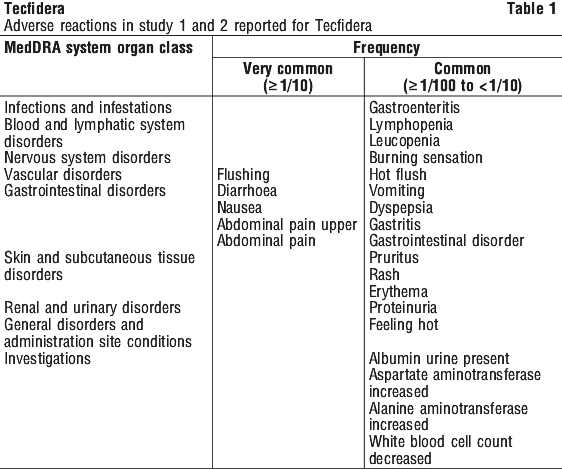
 Other relevant ADRs (< 2% difference) include: gastroenteritis, gastritis, gastrointestinal disorder, burning sensation, feeling hot, alanine aminotransferase increased, proteinuria, white blood cell count decreased and leucopenia.
Other relevant ADRs (< 2% difference) include: gastroenteritis, gastritis, gastrointestinal disorder, burning sensation, feeling hot, alanine aminotransferase increased, proteinuria, white blood cell count decreased and leucopenia. Study 2 (CONFIRM) was a 2 year multicentre, randomised, double blind, placebo controlled study which contained a rater blinded (i.e. study physician/investigator assessing the response to study treatment is blinded) reference comparator of glatiramer acetate (GA) in 1417 patients with RRMS.
Study 2 (CONFIRM) was a 2 year multicentre, randomised, double blind, placebo controlled study which contained a rater blinded (i.e. study physician/investigator assessing the response to study treatment is blinded) reference comparator of glatiramer acetate (GA) in 1417 patients with RRMS. Pooled results at 2 years for study 1 and study 2 showed consistent and statistically significant results for Tecfidera versus placebo in all primary and secondary endpoints, including time to confirmed disability progression (32% relative reduction compared to placebo).
Pooled results at 2 years for study 1 and study 2 showed consistent and statistically significant results for Tecfidera versus placebo in all primary and secondary endpoints, including time to confirmed disability progression (32% relative reduction compared to placebo).

 DMF is a white to off-white powder that is slightly soluble in water. It has a molecular formula of C6H8O4 and a molecular weight of 144.13. The chemical name for DMF is dimethyl (2E)but-2-enedioate.
DMF is a white to off-white powder that is slightly soluble in water. It has a molecular formula of C6H8O4 and a molecular weight of 144.13. The chemical name for DMF is dimethyl (2E)but-2-enedioate.