
What is in this leaflet
This leaflet answers some common questions about FORXIGA. It does not contain all of the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking FORXIGA against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What FORXIGA is used for
FORXIGA is a prescription medicine used with diet, exercise and sometimes other medicines (which may include metformin; insulin; a sulfonylurea medicine such as gliclazide, glimepiride and glibenclamide; or a dipeptidyl peptidase-4 inhibitor [DPP 4 inhibitor] such as sitagliptin or saxagliptin or a glucagon like peptide 1 [GLP-1] receptor agonist such as exenatide) to control the levels of blood sugar (glucose) in adults with type 2 diabetes mellitus. It can also reduce hospitalisation for heart failure in adults with type 2 diabetes. FORXIGA is also used with other medicines to treat heart failure and to slow the progression of kidney disease in adults.
Type 2 diabetes mellitus
Type 2 diabetes mellitus (also called non-insulin-dependent diabetes mellitus, or NIDDM) is the condition of having high levels of blood sugar (hyperglycaemia). This is usually because your body does not make enough insulin and /or the insulin that your body produces does not work as well as it should. Left uncontrolled, the build-up of high levels of sugar in your blood can lead to serious medical problems.
The main goal of treating type 2 diabetes is to control your blood sugar to a normal level. Lowering and controlling blood sugar may help prevent or delay complications of diabetes, which include kidney disease, blindness and amputation.
FORXIGA is a member of a class of medicines you take by mouth called SGLT-2 (Sodium Glucose Cotransporter-2) inhibitors that lower blood sugar levels in patients with type 2 diabetes mellitus.
- FORXIGA lowers the level of your blood sugar by removing the excess sugar from the body in the urine.
- FORXIGA by itself is unlikely to cause low blood sugar (hypoglycaemia) because it does not interfere with the insulin hormone that regulates blood sugar.
Heart failure
Heart failure occurs when the heart is weak and cannot pump enough blood to the lungs and the rest of the body. This can lead to serious medical problems and need for hospital care.
The most common symptoms of heart failure are breathlessness, fatigue, tiredness and ankle swelling.
FORXIGA helps protect your heart from getting weaker and improves your symptoms. It can lower the need to go to hospital.
Kidney disease
Some conditions such as diabetes and high blood pressure can lead to kidney problems. These problems develop slowly over several years. Good control of your blood sugar and blood pressure are important in keeping your kidneys healthy, but may not always prevent kidney damage from occurring.
Your doctor may have prescribed this medicine for another reason. Ask your doctor if you have any questions about why FORXIGA has been prescribed for you.
This medicine is only available with a doctor's prescription.
FORXIGA is not addictive.
Before you take FORXIGA
When you must not take it
Do not take FORXIGA if you have:
- An allergy to dapagliflozin, the active ingredient in FORXIGA or any of the other ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body; rash, itching or hives on the skin or you may feel faint.
Do not take FORXIGA after the use by (expiry) date printed on the pack.
Do not take FORXIGA if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
FORXIGA is not recommended for use in children. It has not been studied in children younger than 18 years old.
If you are not sure whether you should start taking FORXIGA, talk to your doctor or pharmacist.
Before you start to take it
Tell your doctor if you have any allergies to:
- any other medicines
- any other substances such as foods, dyes or preservatives
Tell your doctor or pharmacist if you have, or have had, any medical conditions, especially the following:
- type 1 diabetes mellitus
- kidney, liver or pancreas problems
- you are taking a medicine for high blood pressure or taking a water pill (diuretic)
- frequently get genital or urinary tract infections (infections of the bladder, kidney, or tubes that carry urine)
- an illness that will make you dehydrated such as diarrhoea or a severe infection.
- diabetic ketoacidosis. This is a symptom of uncontrolled diabetes, in which substances called ketone bodies build up in the blood. You may notice this as rapid weight loss, feeling sick or being sick, stomach pain, excessive thirst, fast and deep breathing, confusion, unusual sleepiness or tiredness, a sweet smell to your breath, a sweet or metallic taste in your mouth, or a different odour to your urine or sweat.
Tell your doctor if you are pregnant or intend to become pregnant. The safety of FORXIGA in pregnant women has not been established. FORXIGA must not be used during the second and third trimesters of your pregnancy. If you are pregnant, stop taking FORXIGA and speak with your doctor immediately as good control of your type 2 diabetes (control your blood sugar) is important while you are pregnant and it is not known if FORXIGA will harm your unborn baby.
Tell your doctor if you are breastfeeding or plan to breastfeed. FORXIGA should not be used while breastfeeding or if planning to breastfeed. It is not known if FORXIGA will pass into your breast milk. Talk with your doctor about the best way to feed your baby if you are taking FORXIGA.
Tell your doctor if you are lactose intolerant. FORXIGA tablets contain lactose.
Taking other medicines
Tell your doctor if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food shop.
FORXIGA can be taken with most other medicines.
If you have not told your doctor about any of these things, tell them before you take any FORXIGA.
How to take FORXIGA
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the pack ask your doctor or pharmacist for help.
How to take it
Swallow your FORXIGA tablet with a full glass of water.
How much to take
The dose of FORXIGA is one 10 mg tablet once a day. You should not take more than one FORXIGA tablet per day.
Your doctor may prescribe FORXIGA along with certain other medicines.
When to take it
FORXIGA should be taken once daily at any time of the day regardless of meals.
Taking your tablets at the same time each day will have the best effect. It will also help you remember when to take the tablets.
FORXIGA tablets can be taken with or without food.
How long to take it
Continue taking the tablets for as long as your doctor tells you. Make sure you keep enough FORXIGA to last over weekends and holidays.
FORXIGA helps control your condition, but does not cure it. Therefore, you must take FORXIGA every day.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for the dose that you missed.
If you have trouble remembering when to take your medicine, ask your pharmacist for some hints.
If you take too much
Immediately telephone your doctor or the Poisons Information Centre (13 11 26) for advice, or go to Accident and Emergency at your nearest hospital if you think that you or anyone else may have taken too much FORXIGA. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are taking FORXIGA
Things you must do
Tell any other doctors, dentists, and pharmacists who are treating you that you are taking FORXIGA.
If you are about to be started on any new medicines, tell your doctor, dentist or pharmacist that you are taking FORXIGA.
Make sure that you, your friends, family and work colleagues can recognise the symptoms of hypoglycaemia and hyperglycaemia and know how to treat them.
It is important to have regular check-ups with your doctor or diabetes centre.
If you have diabetes, it is important to check your feet regularly and adhere to any other advice regarding foot care given by your doctor.
Tell your doctor if you experience rapid weight loss, feeling sick or being sick, stomach pain, excessive thirst, fast and deep breathing, confusion, unusual sleepiness or tiredness, a sweet smell to your breath, a sweet or metallic taste in your mouth, or a different odour to your urine or sweat. These symptoms could be a sign of diabetic ketoacidosis.
Talk to your doctor if you are having surgery to discuss when to stop taking FORXIGA and when to start taking it again.
HYPOGLYCAEMIA
FORXIGA does not normally cause hypoglycaemia, although you may experience it if you take certain other medicines, such as insulin or a sulfonylurea.
Hypoglycaemia can occur suddenly. Initial signs may include:
- weakness, trembling or shaking
- sweating
- lightheadedness, dizziness, headache or lack of concentration
- irritability, tearfulness or crying
- hunger
- numbness around the lips and tongue.
If not treated promptly, these may progress to:
- loss of co-ordination
- slurred speech
- confusion
- fits or loss of consciousness.
If you experience any of the symptoms of hypoglycaemia, you need to raise your blood glucose immediately.
You can do this by doing one of the following:
- eating 5 to 7 jelly beans
- eating 3 teaspoons of sugar or honey
- drinking half a can of non-diet soft drink
- taking 2 to 3 concentrated glucose tablets.
Unless you are within 10 to 15 minutes of your next meal or snack, follow up with extra carbohydrates such as plain biscuits, fruit or milk. Taking this extra carbohydrate will prevent a second drop in your blood glucose level.
HYPERGLYCAEMIA
If you notice the return of any of the signs of hyperglycaemia, contact your doctor immediately. Your doctor may need to consider additional or other treatments for your diabetes.
The risk of hyperglycaemia is increased in the following situations:
- uncontrolled diabetes
- illness, infection or stress
- taking less FORXIGA than prescribed
- taking certain other medicines
- too little exercise
- eating more carbohydrates than normal.
Tell your doctor if you:
- become ill
- become dehydrated
- are injured
- have a fever
- have a serious infection
- are having surgery (including dental surgery).
Your blood glucose may become difficult to control at these times.
If you become pregnant while taking FORXIGA, tell your doctor immediately.
If you need to have any medical tests while you are taking FORXIGA, tell your doctor. FORXIGA may affect the results of some tests.
Visit your doctor regularly for check ups. Your doctor may want to perform blood tests to check your kidneys, liver, heart, and levels of cholesterol and fats in your blood while you are taking FORXIGA.
Things you must not do
Do not take FORXIGA to treat any other complaints unless your doctor tells you to.
Do not give this medicine to anyone else, even if their symptoms seem similar or they have the same condition as you.
Things to be careful of
Be careful driving or operating machinery until you know how FORXIGA affects you. Although rare, FORXIGA may cause dizziness in some people. Low blood sugar levels may also slow your reaction time and affect your ability to drive or operate machinery.
Make sure you know how you react to FORXIGA before you drive a car, operate machinery or do anything else that could be dangerous if you are dizzy or lightheaded.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking FORXIGA.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
Side effects of FORXIGA include:
- hypoglycaemia (see below)
- genital infections as suggested by irritation of the genitals
- urinary tract infections
- back pain
- changes in the amount of cholesterol or fats in your blood
- headache
Low blood sugar (hypoglycaemia) may become worse in people who already take another medication to treat diabetes, such as sulfonylureas or insulin. Tell your doctor if you take other diabetes medicines. If you have symptoms of low blood sugar, you should check your blood sugar and treat if low, then call your doctor. Signs of low blood sugar may include weakness, trembling or shaking, sweating, light-headedness, headache, dizziness, rapid heartbeat, lack of concentration, tearfulness or crying, irritability, hunger and numbness around the lips and fingers. Do not drive a car if you have signs of low blood sugar.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital.
- Volume depletion (loss of needed fluids from the body; dehydration). Tell your doctor if you are unable to keep fluids down or if you have any of these symptoms of too much loss of body fluids (volume depletion or dehydration) while taking FORXIGA: dry sticky mouth, severe thirst, severe diarrhoea or vomiting, dizziness, or urinating less often than normal or not at all.
- Shortness of breath, wheezing or severe difficulty in breathing; shock, swelling of the face, lips, tongue or other parts of the body; skin rash, itching or hives on the skin, hayfever, or you may feel faint
- Genital infections. If you take FORXIGA, you may be at a greater risk for genital infections. Tell your doctor if you experience painful urination, soreness and more severe irritation or redness and swelling of your genitals, or an unpleasant odour or discharge associated with your genitals.
- Urinary tract infection. If you take FORXIGA, you may be at a greater risk for urinary tract infections. If you have symptoms, such as burning or pain when you pass urine, more frequent or urgent need to urinate, fever, chills, or blood in the urine, contact your doctor as soon as possible.
- If you experience pain or tenderness, redness, swelling of the genitals or the area from the genitals to the rectum, fever, and generally feeling unwell. These may be symptoms of a rare but serious and potentially life-threating infection called Necrotising fasciitis of the perineum (Fournier's gangrene) and you will require prompt treatment. Your doctor may tell you to stop taking FORXIGA.
- Diabetic ketoacidosis. In rare cases dapagliflozin, the active ingredient in FORXIGA, may cause a serious condition called diabetic ketoacidosis. Symptoms of diabetic ketoacidosis may include feeling sick or being sick, difficulty breathing, severe thirst, feeling weak and tired, confusion, a sweet smell to your breath, a sweet or metallic taste in your mouth, a strange odour to your urine or sweat and frequent urination. The risk of developing diabetic ketoacidosis may be increased with prolonged fasting, excessive alcohol consumption, dehydration, sudden reductions in insulin dose, or a higher need of insulin due to major surgery or serious illness. Diabetes ketoacidosis is a life-threatening condition.
These are very serious side effects. You may need urgent medical attention or hospitalisation.
Other side effects not listed here may occur in some patients. Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
After taking FORXIGA
Storage
Keep your FORXIGA tablets in the blister until it is time to take them. If you take FORXIGA out of the blister it will not keep well.
Keep it in a cool dry place where the temperature stays below 30°C.
Do not store it or any other medicine in the bathroom or near a sink. Do not leave it in the car or on a window sill. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking FORXIGA or the tablets have passed their expiry date, ask your pharmacist what to do with any that are left over.
Product description
What FORXIGA looks like
FORXIGA tablets are available in one strength i.e. 10 mg. The tablets are film-coated, yellow, biconvex and diamond-shaped with "10" engraved on one side and "1428" engraved on the other side.
FORXIGA tablets are available in blister packs of 7 and 28.
Ingredients
Each FORXIGA tablet contains dapagliflozin 10 mg as the active ingredient.
Plus the following inactive ingredients:
- microcrystalline cellulose
- lactose
- crospovidone
- silicon dioxide
- magnesium stearate
- polyvinyl alcohol
- titanium dioxide
- macrogol 3350
- purified talc
- iron oxide yellow
FORXIGA tablets do not contain gluten or sucrose.
Sponsored by
AstraZeneca Pty Ltd
ABN 54 009 682 311
66 Talavera Road
MACQUARIE PARK NSW 2113
Telephone: 1800 805 342
This leaflet was prepared:
September 2021
FORXIGA Australian Registration Number:
10 mg - AUST R 180147
FORXIGA is a registered trademark of the AstraZeneca group of companies.
Doc ID-002678542 v15.0
Published by MIMS October 2021
 Additional adverse reactions in ≥ 5% of patients treated with Forxiga 10 mg, ≥ 1% more than patients in placebo/ comparator, and reported in at least three more patients treated with Forxiga 10 mg and regardless of relationship to Forxiga reported by investigator, are described below by treatment regimen.
Additional adverse reactions in ≥ 5% of patients treated with Forxiga 10 mg, ≥ 1% more than patients in placebo/ comparator, and reported in at least three more patients treated with Forxiga 10 mg and regardless of relationship to Forxiga reported by investigator, are described below by treatment regimen.


 Forxiga as an add-on with either metformin, glimepiride, metformin and a sulfonylurea, sitagliptin (with or without metformin), saxagliptin with metformin or insulin resulted in statistically significant reductions in HbA1c at 24 weeks compared with subjects receiving placebo (p < 0.0001; see Tables 5, 6 and 7).
Forxiga as an add-on with either metformin, glimepiride, metformin and a sulfonylurea, sitagliptin (with or without metformin), saxagliptin with metformin or insulin resulted in statistically significant reductions in HbA1c at 24 weeks compared with subjects receiving placebo (p < 0.0001; see Tables 5, 6 and 7).

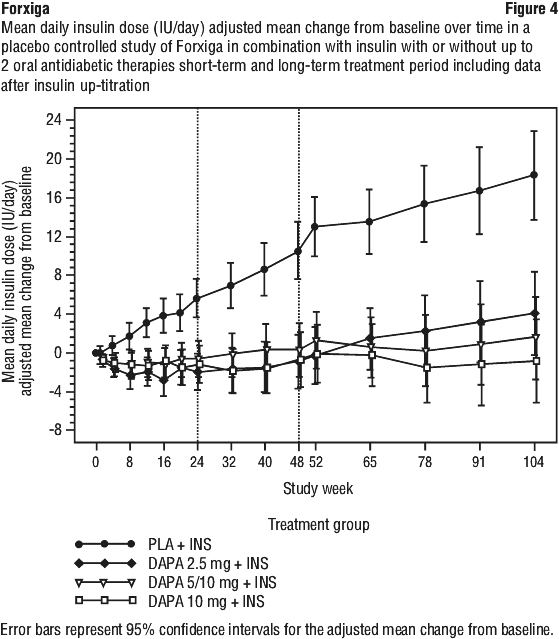
 The reductions in HbA1c observed at week 24 were sustained in add-on combination studies (glimepiride and insulin) with 48 week data (glimepiride) and up to 104 week data (insulin, see Figure 2). At week 48 when added to sitagliptin (with or without metformin), the adjusted mean change from baseline for Forxiga 10 mg and placebo was -0.30% and 0.38%, respectively. For the add-on to metformin study, HbA1c reductions were sustained through week 102 (-0.78% and 0.02% adjusted mean change from baseline for 10 mg and placebo, respectively, also see Figure 3). At week 104 for insulin (with or without additional oral glucose lowering medicinal products), the HbA1c reductions were -0.71% and -0.06% adjusted mean change from baseline for Forxiga 10 mg and placebo, respectively. At weeks 48 and 104, the insulin dose remained stable compared to baseline in subjects treated with Forxiga 10 mg at an average dose of 76 IU/day (see Figure 4). In the placebo group there was a mean increase of 10.5 IU/day and 18.3 IU/day from baseline at weeks 48 and 104, respectively. The proportion of subjects remaining in the study at week 104 was 72.4% for the group treated with Forxiga 10 mg and 54.8% for the placebo group.
The reductions in HbA1c observed at week 24 were sustained in add-on combination studies (glimepiride and insulin) with 48 week data (glimepiride) and up to 104 week data (insulin, see Figure 2). At week 48 when added to sitagliptin (with or without metformin), the adjusted mean change from baseline for Forxiga 10 mg and placebo was -0.30% and 0.38%, respectively. For the add-on to metformin study, HbA1c reductions were sustained through week 102 (-0.78% and 0.02% adjusted mean change from baseline for 10 mg and placebo, respectively, also see Figure 3). At week 104 for insulin (with or without additional oral glucose lowering medicinal products), the HbA1c reductions were -0.71% and -0.06% adjusted mean change from baseline for Forxiga 10 mg and placebo, respectively. At weeks 48 and 104, the insulin dose remained stable compared to baseline in subjects treated with Forxiga 10 mg at an average dose of 76 IU/day (see Figure 4). In the placebo group there was a mean increase of 10.5 IU/day and 18.3 IU/day from baseline at weeks 48 and 104, respectively. The proportion of subjects remaining in the study at week 104 was 72.4% for the group treated with Forxiga 10 mg and 54.8% for the placebo group.







 When evaluating the renal components, there were 127 and 238 events of new or worsening nephropathy (sustained eGFR decrease, ESKD or renal death) in patients in the Forxiga and placebo groups, respectively. The HR for time to nephropathy was 0.53 (95% CI 0.43, 0.66) for Forxiga versus placebo.
When evaluating the renal components, there were 127 and 238 events of new or worsening nephropathy (sustained eGFR decrease, ESKD or renal death) in patients in the Forxiga and placebo groups, respectively. The HR for time to nephropathy was 0.53 (95% CI 0.43, 0.66) for Forxiga versus placebo. The safety profile of dapagliflozin in the study was consistent with that in the general population of patients with type 2 diabetes. Mean eGFR decreased initially during the treatment period in the dapagliflozin group and subsequently remained stable during the 24 week treatment period (Forxiga: -3.39 mL/min/1.73 m2 and placebo: 0.90 mL/min/1.73 m2). At 3 weeks after termination of Forxiga, the mean change from baseline in eGFR in the dapagliflozin group was similar to the mean change in the placebo group (Forxiga: 0.57 mL/min/1.73 m2 and placebo: -0.04 mL/min/1.73 m2).
The safety profile of dapagliflozin in the study was consistent with that in the general population of patients with type 2 diabetes. Mean eGFR decreased initially during the treatment period in the dapagliflozin group and subsequently remained stable during the 24 week treatment period (Forxiga: -3.39 mL/min/1.73 m2 and placebo: 0.90 mL/min/1.73 m2). At 3 weeks after termination of Forxiga, the mean change from baseline in eGFR in the dapagliflozin group was similar to the mean change in the placebo group (Forxiga: 0.57 mL/min/1.73 m2 and placebo: -0.04 mL/min/1.73 m2). All three components of the primary composite endpoint individually contributed to the treatment effect (Figure 12). There were few urgent heart failure visits. Forxiga also reduced the incidence of cardiovascular death or hospitalization for heart failure (HR 0.75 [95% CI 0.65, 0.85], p < 0.0001).
All three components of the primary composite endpoint individually contributed to the treatment effect (Figure 12). There were few urgent heart failure visits. Forxiga also reduced the incidence of cardiovascular death or hospitalization for heart failure (HR 0.75 [95% CI 0.65, 0.85], p < 0.0001). Forxiga also reduced the total number of events of hospitalizations for heart failure (first and recurrent) and cardiovascular death; there were 567 events in the Forxiga group versus 742 events in the placebo group (Rate Ratio 0.75 [95% CI 0.65, 0.88]; p=0.0002).
Forxiga also reduced the total number of events of hospitalizations for heart failure (first and recurrent) and cardiovascular death; there were 567 events in the Forxiga group versus 742 events in the placebo group (Rate Ratio 0.75 [95% CI 0.65, 0.88]; p=0.0002). The treatment benefit of Forxiga over placebo on the primary endpoint was also consistent across other key subgroups (Figure 14).
The treatment benefit of Forxiga over placebo on the primary endpoint was also consistent across other key subgroups (Figure 14).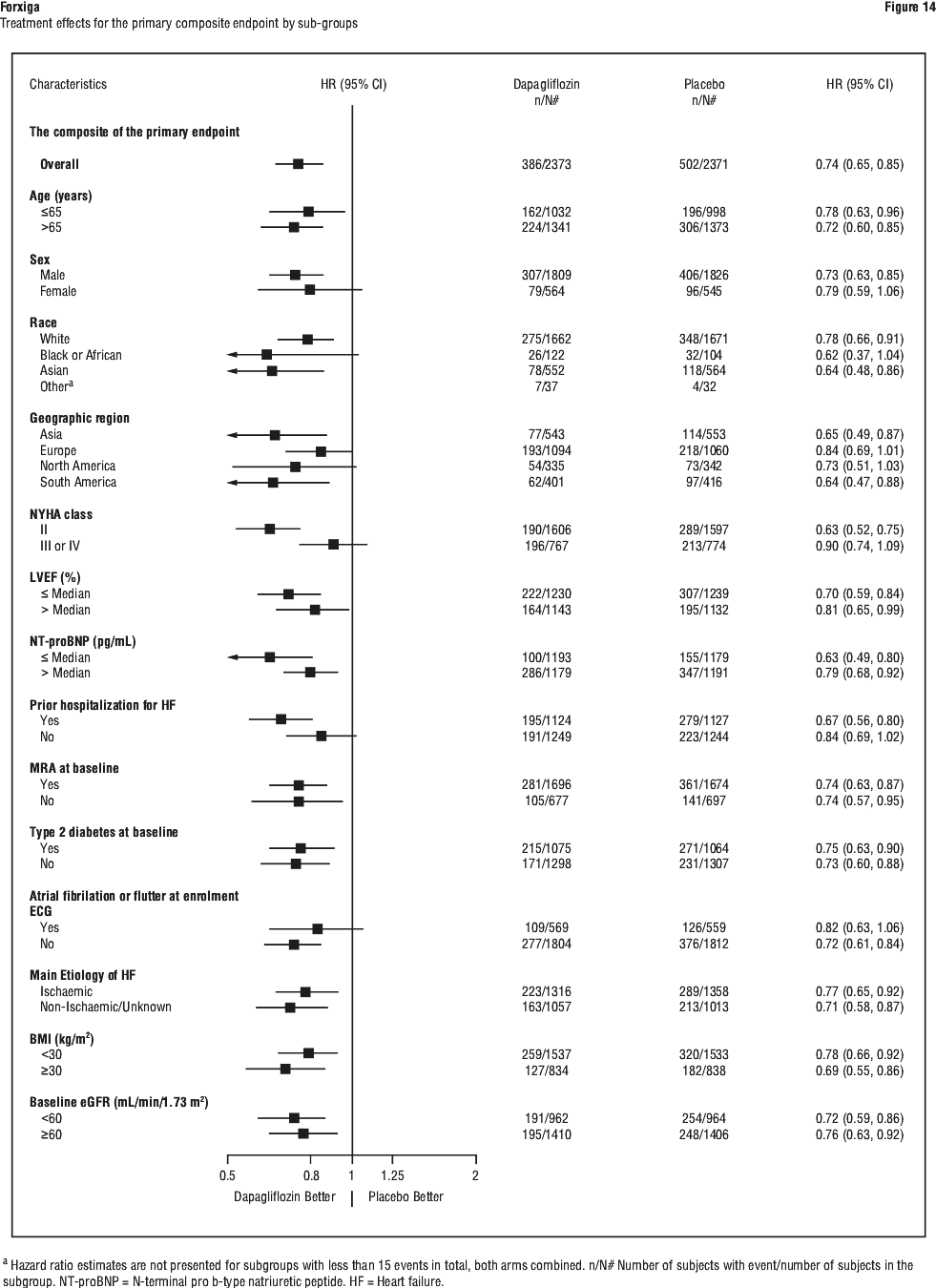

 All three components of the primary composite endpoint individually contributed to the treatment effect (Figure 16).
All three components of the primary composite endpoint individually contributed to the treatment effect (Figure 16). Forxiga was superior to placebo in reducing the rate of the composite of cardiovascular death and total number of heart failure events (first and recurrent hospitalization for heart failure or urgent heart failure visits); there were 815 events in the Forxiga group versus 1057 events in the placebo group (rate ratio 0.77 [95% CI 0.67, 0.89]; p=0.0003).
Forxiga was superior to placebo in reducing the rate of the composite of cardiovascular death and total number of heart failure events (first and recurrent hospitalization for heart failure or urgent heart failure visits); there were 815 events in the Forxiga group versus 1057 events in the placebo group (rate ratio 0.77 [95% CI 0.67, 0.89]; p=0.0003).

 All four components of the primary composite endpoint individually contributed to the treatment effect (Figure 19). Forxiga also reduced the incidence of the composite endpoint of ≥ 50% sustained decline in eGFR, ESKD or renal death (HR 0.56 [95% CI 0.45, 0.68], p < 0.0001), the composite endpoint of CV death and hospitalization for heart failure (HR 0.71 [95% CI 0.55, 0.92], p = 0.0089), and all-cause mortality (HR 0.69 [95% CI 0.53, 0.88], p = 0.0035) (Figure 20).
All four components of the primary composite endpoint individually contributed to the treatment effect (Figure 19). Forxiga also reduced the incidence of the composite endpoint of ≥ 50% sustained decline in eGFR, ESKD or renal death (HR 0.56 [95% CI 0.45, 0.68], p < 0.0001), the composite endpoint of CV death and hospitalization for heart failure (HR 0.71 [95% CI 0.55, 0.92], p = 0.0089), and all-cause mortality (HR 0.69 [95% CI 0.53, 0.88], p = 0.0035) (Figure 20).
 The treatment effect of Forxiga was consistent in chronic kidney disease patients with type 2 diabetes mellitus and without diabetes (Figure 21).
The treatment effect of Forxiga was consistent in chronic kidney disease patients with type 2 diabetes mellitus and without diabetes (Figure 21). The treatment benefit of Forxiga over placebo on the primary composite endpoint was consistent across key subgroups (Figure 22).
The treatment benefit of Forxiga over placebo on the primary composite endpoint was consistent across key subgroups (Figure 22). There were fewer patients with renal-related adverse events in the Forxiga group compared with the placebo group, 144 (6.7%) and 169 (7.9%) in the Forxiga and placebo groups, respectively.
There were fewer patients with renal-related adverse events in the Forxiga group compared with the placebo group, 144 (6.7%) and 169 (7.9%) in the Forxiga and placebo groups, respectively. Molecular formula: C21H25ClO6.C3H8O2.H2O.
Molecular formula: C21H25ClO6.C3H8O2.H2O.