

What is in this leaflet
This leaflet answers some common questions about LOGEM. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking LOGEM against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, talk to your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What LOGEM is used for
LOGEM contains lamotrigine as the active ingredient and belongs to a group of medicines called "anti-epileptic drugs".
Anti-epileptic drugs, such as LOGEM, are used to treat epilepsy in adults and children aged 2 years and older. Epilepsy is a condition where you have repeated seizures (fits).
Initially, LOGEM is usually used in addition to other medicines for the treatment of epilepsy including partial or generalised seizures and Lennox-Gastaut Syndrome.
It is thought that this medicine works by changing the levels of some chemicals associated with seizures.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is only available with a doctor's prescription.
This medicine is not addictive.
Before you take LOGEM
When you must not take it
Do not take LOGEM if you have an allergy to:
- any medicine containing lamotrigine
- any of the ingredients listed at the end of this leaflet
Symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty in breathing
- swelling of the face, lips, tongue or any other parts of the body
- rash, itching or hives on the skin
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damage, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- a history of allergy or rash to other anti-epileptic drugs
- liver or kidney disorders
- Parkinson's disease
- if you have ever developed meningitis after taking lamotrigine
Tell your doctor if you are pregnant or plan to become pregnant. LOGEM may affect your unborn baby if you take it during pregnancy but it is still important that you control your fits whilst you are pregnant. Your doctor will discuss the risks and benefits of taking this medicine during pregnancy.
It is recommended that women on anti-epileptic drugs, such as LOGEM, receive pre-pregnancy counselling with regard to the possible risk to their unborn child.
Studies have shown a decrease in the levels of folic acid during pregnancy when LOGEM is also used. It is therefore recommended that you take a daily 5 mg folate supplement before becoming pregnant and during the first 12 weeks of your pregnancy.
Tell your doctor if you are breast feeding or planning to breast feed. LOGEM can pass into breast milk and may affect your baby. Your doctor will discuss the risks and benefits of taking this medicine if you are breast feeding.
If you have not told your doctor about any of the above, tell them before you start taking LOGEM.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from a pharmacy, supermarket or health food shop.
Some medicines may interfere with LOGEM. These include:
- valproate and carbamazepine, used to treat both epilepsy and mental health problems
- any form of hormonal medicine, e.g. "the pill" or HRT
- other anti-epileptic drugs, e.g. phenytoin, primidone or phenobarbitone
- OCT2 substrates such as dofetilide
- rifampicin, an antibiotic, which is used to treat infections, including tuberculosis
- medicines used to treat Human Immunodeficiency Virus (HIV) infection
- risperidone, used to treat mental health problems
These medicines may be affected by LOGEM or may affect how well it works. You may need different amounts of your medicines or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take LOGEM
Follow all directions given to you by your doctor and pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
Using LOGEM for the first time
You may notice that you feel dizzy, tired or unsteady in the first few weeks of treatment with LOGEM tablets. During this period you may also notice that you have slight problems with your vision. As your reactions may be slower during this period you should not operate any machinery or drive a car. If any of these effects do not go away or are troublesome you should see your doctor.
Tell your doctor immediately if you develop any skin rash such as spots or 'hives' during LOGEM treatment. There are reports of severe, potentially life-threatening rashes associated with LOGEM treatment, particularly in children. LOGEM should be discontinued at the first sign of rash unless the rash is clearly not drug related.
Contact your doctor if you experience a rash or sunburn after taking Lamictal and having been exposed to sun or artificial light, e.g. solarium. Your doctor will check your treatment and may advise you to avoid sunlight or protect yourself against the sun, e.g. use of a sunscreen and/or to wear protective clothing.
If you have any questions about taking LOGEM ask your doctor or pharmacist.
How much to take
It may take a while to find the best dose of LOGEM for you.
The dose you take will depend on:
- your age and weight
- whether you are taking LOGEM with other medications
- whether you have any kidney or liver problems
Never take more of this medicine than your doctor tells you to.
Do not increase the dose faster than you have been told. Your doctor will prescribe a low dose to start and gradually increase the dose over a few weeks until you reach a dose that works for you.
Women taking hormonal contraceptives, such as the birth control pill may need a higher maintenance dose of LOGEM. Your doctor will usually decrease your dose once you stop taking hormonal contraceptives.
Tell your doctor if there are any changes in your menstrual pattern, such as breakthrough bleeding whilst on "the pill". Your doctor may need to change the dose of LOGEM as "the pill" may not work as effectively for contraception whilst taking it.
How to take it
LOGEM tablets may be swallowed whole, chewed or dispersed in sufficient water to cover the whole tablet.
LOGEM can be taken with or without food.
Your doctor may also advise you to start or stop taking other medications, depending on what condition you are being treated for and the way you respond to treatment.
How long to take it
Continue taking your medicine for as long as your doctor tells you.
This medicine helps to control your condition, but does not cure it. It is important to keep taking your medicine even if you feel well.
Do not stop taking LOGEM or change the dose without first checking with your doctor. Your doctor will advise you if you need to stop taking LOGEM and how to do this gradually over a period of two weeks.
Use in children
LOGEM is not recommended for treatment of epilepsy in children under 2 years of age. Children's weight should be checked and the dose reviewed as weight changes with growth occur.
If you forget to take it
If you have forgotten to take your dose of LOGEM contact your doctor immediately.
Do not take a double dose to make up for the dose you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much LOGEM. Do this even if there are no signs of discomfort or poisoning.
If you take too much Lamictal you may be more likely to have serious side effects which may be fatal.
You may need urgent medical attention.
Symptoms of LOGEM overdose can include rapid, uncontrollable eye movements, clumsiness and lack of coordination affecting your balance, impaired or loss of consciousness, fits or coma.
While you are taking LOGEM
Your doctor or pharmacist will be able to tell you whether there are any special instructions while you are taking LOGEM tablets.
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking LOGEM.
Tell any other doctors, dentists and pharmacists who treat you that you are taking this medicine, you are about to be started on any new medicines.
Take LOGEM exactly as your doctor has prescribed.
If you develop any skin rash (e.g. spots or 'hives') during LOGEM treatment contact your doctor immediately. There are reports of severe, potentially life-threatening rashes associated with LOGEM treatment, particularly in children. LOGEM should be discontinued at the first sign of rash unless the rash is clearly not drug related.
Contact your doctor if you experience a rash or sunburn after taking Lamictal and having been exposed to sun or artificial light, e.g. solarium. Your doctor will check your treatment and may advise you to avoid sunlight or protect yourself against the sun, e.g. use of a sunscreen and/or to wear protective clothing.
Seek immediate medical attention if you experience symptoms such as fever, rash or enlarged/swollen lymph nodes whilst taking this medicine.
If you are about to have a blood test, tell your doctor or hospital that you are taking this medicine. LOGEM may interfere with some laboratory tests to detect other drugs.
Talk to your doctor as soon as possible if you are pregnant or if you are planning to become pregnant.
Your doctor will discuss the risks and benefits of taking LOGEM during pregnancy.
Talk to your doctor if you are breast feeding or planning to breast feed.
LOGEM can pass into breast milk and may affect your baby. Your doctor will discuss the risks and benefits of breast feeding while you are taking it.
Keep all of your doctor's appointments so that your progress can be checked.
Tell your doctor if, for any reason, you have not taken your medicine exactly as directed.
Otherwise, your doctor may think that it was not working as it should and change your treatment unnecessarily.
Things you must not do
Do not take LOGEM to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or change the dose without checking with your doctor.
Do not take a double dose to make up for any that you may have missed.
If you stop taking it suddenly your epilepsy may return or become worse. This is known as "rebound seizures".
Your doctor will advise you if you need to stop taking LOGEM and how to do this gradually over about 2 weeks.
Things to be careful of
Be careful driving or operating machinery until you know how LOGEM affects you.
As with other anticonvulsant medicines for the treatment of epilepsy, LOGEM may cause dizziness and drowsiness in some people and affect alertness.
Make sure you know how you react to LOGEM before you drive a car, operate machinery or do anything else that could be dangerous if you are dizzy or light-headed. If this occurs do not drive.
Children should not ride a bike, climb trees or do anything else that could be dangerous if they are feeling dizzy or sleepy.
Tell your doctor immediately or go to the Accident and Emergency department of your nearest hospital if you or someone you know has any suicidal thoughts or other mental/mood changes. All mentions of suicide or violence must be taken seriously. Families and caregivers of children and adolescents who are taking LOGEM should be especially watchful for any changes in behaviour. Anti-epileptic medicines such as LOGEM may increase the risk of suicidal behaviour (including suicidal thoughts and suicide attempts).
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking LOGEM.
This medicine helps most people with epilepsy, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you have some of the side effects.
Do not be alarmed by the following list of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
The most commonly reported side effects of LOGEM are:
- dizziness
- movement problems such as tics, unsteadiness and jerkiness
- tremors
- skin rash
- headache
- nausea
- vomiting
- feeling drowsy or tired
- blurred or double vision
- rapid, uncontrollable eye movements
- trouble sleeping
- feeling sleepy
- irritability, aggression or agitation
- hallucinations, confusion
- increased activity in children
- joint, back or stomach pain
- respiratory or lung problems
- depression
- loss of memory
- liver problems
- diarrhoea
- dry mouth
In general, these side effects usually happen during the first few weeks of treatment with LOGEM.
Tell your doctor immediately or go to the Accident and Emergency department of your nearest hospital if you or someone you know has any suicidal thoughts or other mental/mood changes whilst taking LOGEM. All mentions of suicide or violence must be taken seriously. Families and caregivers of children and adolescents who are taking LOGEM should be especially watchful for any changing behaviour. Anti-epileptic medicines such as LOGEM may increase the risk of suicidal behaviour (including suicidal thoughts and suicide attempts).
Potentially serious skin reaction
A small number of people taking LOGEM get an allergic reaction or potentially serious skin reaction, which may develop into more serious problems if they are not treated. Severe allergic reactions are rare.
These symptoms are more likely to happen during the first few months of treatment with LOGEM, especially if the dose is too high or if the dose is increased too quickly, or if LOGEM is taken with another medicine called valproate. Serious skin reactions are more common in children.
Symptoms of these serious allergic reactions include:
- any skin reaction (e.g. rash or 'hives')
- skin rash or sunburn after exposure to sun or artificial light (photosensitivity)wheezing, difficulty in breathing
- swelling of the face, lips or tongue
- sore mouth or sore eyes
- fever
- swollen glands
Tell your doctor immediately if you notice any of the above symptoms.
Liver and blood problems
Tell your doctor if you notice any of these symptoms:
- drowsiness
- itching
- abdominal pain or tenderness
- feeling very tired
- unusual bleeding or bruising more easily than normal
- a sore throat or more infections, such as a cold, than usual
- yellowing of the skin (jaundice)
Your doctor may decide to carry out tests on your liver, kidneys or blood and may tell you to stop taking LOGEM if you experience these rare symptoms.
Potentially serious aseptic meningitis
LOGEM increases the risk of meningitis, which is a serious inflammation of the protective membrane that covers the brain and spinal cord.
Many of the side effects already listed are symptoms of this condition, as well as light sensitivity, stiff neck, muscle pains and chills.
Potentially serious / life threatening arrhythmia
If you have had a fast heartbeat, heart failure, or other heart problems, you should not take lamotrigine. This drug may cause you to have anabnormal heartbeat, which could lead to sudden death. Symptoms include a fast, slow, or pounding heartbeat, shortness of breath, chest pain, and feeling lightheaded.
Tell your doctor if you notice any of these symptoms.
Tell your doctor as soon as possible if your seizures get worse or if you have a new type of seizure. You may need urgent medical attention or hospitalisation. Serious side effects are rare.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell, even if you think the problems are not connected with this medicine and are not referred to in this leaflet.
Other side effects not listed above may also occur in some people.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
After taking LOGEM
Storage
Keep your tablets in the pack until it is time to take them. If you take the tablets out of the pack they will not keep well.
Keep your tablets in a cool dry place where the temperature stays below 25°C.
Do not store LOGEM or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep LOGEM where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any that is left over.
Product description
What it looks like
LOGEM is available in 4 strengths:
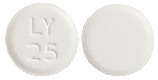
- LOGEM 25 - white to off-white, round, flat faced, bevelled edge tablets with "LY" over "25" on one side and plain on the other side
- LOGEM 50 - white to off-white, round, flat faced, bevelled edge tablets with "LY" over "50" on one side and plain on the other side
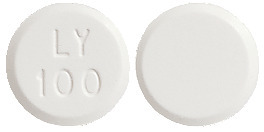
- LOGEM 100 - white to off-white, round, flat faced, bevelled edge tablets with "LY" over "100" on one side and plain on the other side
- LOGEM 200 - white to off-white, round, flat faced, bevelled edge tablets with "LY" over "200" on one side and plain on the other side
LOGEM tablets are available in packs of 56 tablets.
Ingredients
The active ingredient in LOGEM is lamotrigine.
Each LOGEM 25 tablet contains 25 mg of lamotrigine.
Each LOGEM 50 tablet contains 50 mg of lamotrigine.
Each LOGEM 100 tablet contains 100 mg of lamotrigine.
Each LOGEM 200 tablet contains 200 mg of lamotrigine.
The tablets also contain the following inactive ingredients:
- cellulose - microcrystalline
- sodium starch glycollate
- povidone
- purified water
- silicon dioxide
- saccharin sodium
- artificial blackcurrant flavour
- mannitol
- magnesium stearate.
LOGEM contain sulfites. LOGEM tablets do not contain lactose or gluten.
Manufacturer/ Supplier
Alphapharm Pty Ltd trading as Viatris
Level 1, 30 The Bond
30 - 34 Hickson Road
Millers Point NSW 2000
www.viatris.com.au
Phone: 1800 274 276
This leaflet was revised in November 2023
Australian registration numbers:
LOGEM 25 - AUST R 99059
LOGEM 50 - AUST R 99360
LOGEM 100 - AUST R 99062
LOGEM 200 - AUST R 99064
LOGEM® is a Viatris company trade mark.
LOGEM_cmi\Nov 23/00
Published by MIMS December 2023
 Because of a risk of rash, the initial dose and subsequent dose escalation should not be exceeded (see Section 4.4 Special Warnings and Precautions for Use).
Because of a risk of rash, the initial dose and subsequent dose escalation should not be exceeded (see Section 4.4 Special Warnings and Precautions for Use). Because of a risk of rash, the initial dose and subsequent dose escalation should not be exceeded (see Section 4.4 Special Warnings and Precautions for Use).
Because of a risk of rash, the initial dose and subsequent dose escalation should not be exceeded (see Section 4.4 Special Warnings and Precautions for Use). The relative risk for suicidal thoughts or behaviour was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.
The relative risk for suicidal thoughts or behaviour was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications. Approximately 96% of a given dose of lamotrigine is eliminated by conjugation metabolism mediated by glucuronyl transferases. Cytochrome P450 is not involved in the elimination of lamotrigine to any significant extent. Therefore, the likelihood that lamotrigine inhibits the elimination of drugs metabolised by cytochrome P450 is low.
Approximately 96% of a given dose of lamotrigine is eliminated by conjugation metabolism mediated by glucuronyl transferases. Cytochrome P450 is not involved in the elimination of lamotrigine to any significant extent. Therefore, the likelihood that lamotrigine inhibits the elimination of drugs metabolised by cytochrome P450 is low.
 Chemical name: 3,5-diamino-6- (2,3-dichlorophenyl)- 1,2,4-triazine.
Chemical name: 3,5-diamino-6- (2,3-dichlorophenyl)- 1,2,4-triazine.