

SUMMARY CMI
SIMPRAL®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I using SIMPRAL?
SIMPRAL contains the active ingredient pramipexole dihydrochloride monohydrate. SIMPRAL is used to treat the symptoms of Parkinson's disease and Restless Legs Syndrome (RLS).
For more information, see Section 1. Why am I using SIMPRAL? in the full CMI.
2. What should I know before I use SIMPRAL?
Do not use if you have ever had an allergic reaction to SIMPRAL or any of the ingredients listed at the end of the CMI.
Do not give this medicine to a child or adolescent under the age of 18 years.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use SIMPRAL? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with SIMPRAL and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use SIMPRAL?
- Follow all directions given to you by your doctor and pharmacist carefully.
- If you do not understand the instructions on the label, ask your doctor or pharmacist for help.
More instructions can be found in Section 4. How do I use SIMPRAL? in the full CMI.
5. What should I know while using SIMPRAL?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Drinking alcohol |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using SIMPRAL? in the full CMI.
6. Are there any side effects?
SIMPRAL may cause nausea, vomiting, constipation, loss of memory, fainting, signs of allergy, excessive sleepiness.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
FULL CMI
SIMPRAL®
Active ingredient(s): pramipexole dihydrochloride monohydrate
Consumer Medicine Information (CMI)
This leaflet provides important information about using SIMPRAL. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using SIMPRAL.
Where to find information in this leaflet:
1. Why am I using SIMPRAL?
2. What should I know before I use SIMPRAL?
3. What if I am taking other medicines?
4. How do I use SIMPRAL?
5. What should I know while using SIMPRAL?
6. Are there any side effects?
7. Product details
1. Why am I using SIMPRAL?
SIMPRAL contains the active ingredient pramipexole dihydrochloride monohydrate. SIMPRAL belongs to a group of medicines known as "dopamine agonists", which bind to dopamine receptors. SIMPRAL works by having a similar effect as dopamine in the brain.
SIMPRAL is used to treat symptoms of:
- PARKINSON'S DISEASE
- Parkinson's disease is a disease of the brain that affects body movement.
- The symptoms of Parkinson's disease are caused by a lack of dopamine, a naturally occurring chemical produced by certain brain cells.
- Dopamine relays messages in the part of the brain that controls movement. When too little dopamine is produced, this results in Parkinson's disease. - RESTLESS LEGS SYNDROME (RLS)
- RLS is a neurological disorder in which there is an overwhelming urge to move the legs to stop unpleasant sensations.
- The sensations vary from person to person and range from uncomfortable to irritating to painful. The symptoms usually occur when sitting or lying down - which often leads to problems falling or staying asleep. Sometimes the arms and body may be affected. Current evidence suggests that RLS may be due to faulty dopamine signals in certain areas of the brain.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason. This medicine is available only with a doctor's prescription.
SIMPRAL is not addictive.
2. What should I know before I use SIMPRAL?
Warnings
Do not use SIMPRAL:
- if you are allergic to pramipexole dihydrochloride monohydrate, or any of the ingredients listed at the end of this leaflet. Always check the ingredients to make sure you can use this medicine. Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin - in a child or adolescent under the age of 18 years. Safety and effectiveness in children younger than 18 years have not been established.
- after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Check with your doctor if you:
- have allergies to any other medicines, foods, preservatives or dyes.
- have or have had any of the following medical conditions:
- kidney problems
- mental illnesses
- low blood pressure
- trouble controlling your muscles (dyskinesia). - take any medicines for any other condition.
If you have not told your doctor about any of the above, tell him/her before you start taking SIMPRAL.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Check with your doctor if you are pregnant or intend to become pregnant. Your doctor can discuss with you the risks and benefits involved.
Talk to your doctor if you are breastfeeding or intend to breastfeed. SIMPRAL is not recommended during breastfeeding, as it may pass into breast milk.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and SIMPRAL may interfere with each other. These include:
- levodopa, levodopa/carbidopa combination, or other medicines used to treat Parkinson's disease (e.g. amantadine)
- medicines used in the treatment of high blood pressure or heart problems (e.g. digoxin, diltiazem, procainamide, quinidine, triamterene, verapamil, hydrochlorothiazide)
- medicines used in the treatment of mental illness/ psychosis (antipsychotics or neuroleptics)
- metoclopramide, a medicine commonly used to help control nausea and vomiting
- cimetidine or ranitidine, medicines used to treat stomach ulcer or duodenal ulcers
- quinine, a medicine used to treat malaria
- some antibiotics (e.g. trimethoprim, cephalosporins, penicillins)
- indometacin, a medicine used to treat arthritis
- chlorpropamide, a medicine used to treat diabetes
- other medicines that can cause drowsiness or sleepiness (e.g. antihistamine or some cough and cold preparations).
These medicines may be affected by SIMPRAL or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect SIMPRAL.
4. How do I use SIMPRAL?
Follow all directions given to you by your doctor and pharmacist carefully. They may differ from the information contained in this leaflet.
Your doctor or pharmacist will tell you how many tablets you will need to take each day. This depends on your condition and whether or not you are taking any other medicines.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
How much to take
The dose varies from patient to patient. Your doctor may first start you on a low dose of SIMPRAL and slowly increase the amount of SIMPRAL until the right dose is reached to control your condition.
PARKINSON'S DISEASE
- The usual starting dose is one SIMPRAL 0.125 mg tablet three times a day.
- Depending on how you respond to the treatment, your doctor may increase your daily dose gradually in steps of 0.75 mg at weekly intervals until the right dose for your needs is reached. The maximum dose is 4.5 mg of SIMPRAL per day.
RESTLESS LEGS SYNDROME
- The usual starting dose is one SIMPRAL 0.125 mg tablet once a day, usually 2 to 3 hours before you go to bed.
- Depending on how you respond to the treatment, your doctor may increase your dose gradually every 4 to 7 days until the right dose for your needs is reached. The maximum dose is 0.75 mg of SIMPRAL per day.
Follow the instructions provided by your doctor or pharmacist carefully.
When to take SIMPRAL
- SIMPRAL should be used at about the same time each day.
- Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
- It does not matter if you take this medicine before or after food.
How to take SIMPRAL
Swallow the tablets whole with a full glass of water.
How long to take it
Continue taking your medicine for as long as your doctor tells you to.
This medicine helps to control your condition, but does not cure it. It is important to keep taking your medicine even if you feel well.
If you forget to use SIMPRAL
PARKINSON'S DISEASE
- If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
- Otherwise, take it as soon as you remember, and then go back to taking your medicine as you would normally.
- Do not take a double dose to make up for the dose you missed.
- This may increase the chance of you getting an unwanted side effect.
RESTLESS LEGS SYNDROME
- If you forget to take SIMPRAL before you go to bed and you wake up late in the night or early morning, do not take any SIMPRAL as you may have trouble waking in the morning.
- Skip the dose you missed and take the next dose when you are meant to.
- If you are not sure what to do, ask your doctor or pharmacist.
- If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you use too much SIMPRAL
If you think that you have used too much SIMPRAL, you may need urgent medical attention.
Symptoms of an overdose may include nausea, vomiting, abnormal uncontrolled movements, hallucinations, agitation and dizziness or light-headedness.
You should immediately:
- phone the Poisons Information Centre
(Australia telephone 13 11 26) for advice, or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while using SIMPRAL?
Things you should do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking SIMPRAL.
Remind any doctor, dentist or pharmacist you visit that you are using SIMPRAL.
If you feel that SIMPRAL is not helping your condition, tell your doctor or pharmacist. Some people taking long-term SIMPRAL treatment for Restless Legs Syndrome may experience more intense, spreading, or worsening symptoms.
Tell your doctor if, for any reason, you have not used SIMPRAL exactly as prescribed. Otherwise, your doctor may think that it was not effective and change your treatment unnecessarily.
Tell your doctor as soon as possible if there is any worsening of your condition.
Tell your doctor if you experience symptoms such as depression, apathy, anxiety, fatigue, sweating or pain after stopping or reducing your SIMPRAL treatment. If the problems persist for more than a few weeks, your doctor may need to adjust your treatment.
Tell your doctor if you develop an inability to keep your body and neck straight and upright. For example, you may experience abnormal posture such as forward bending of the head and neck, forward bending of the lower back or sidewards bending of the back.
Call your doctor straight away if you or your family notice an increase in compulsive behaviour.
Things you should not do
- Do not stop using this medicine suddenly, unless advised to do so by your doctor.
- If you stop taking it suddenly, your condition may worsen or you may have unwanted side effects.
- If you are using SIMPRAL for your Parkinson's Disease and your doctor asks you to stop taking SIMPRAL, the dose will normally be reduced gradually over several days. - Do not take SIMPRAL to treat any other complaints unless your doctor tells you to.
- Do not give your medicine to anyone else, even if they have the same condition as you.
- Do not stop taking your medicine or change the dosage without checking with your doctor.
Things to be careful of
- Make sure you know how you react to SIMPRAL before you engage in any activities where impaired alertness may put yourself or others at risk of serious injury.
- Be careful getting up from a sitting or lying position. You may feel dizzy or lightheaded while taking SIMPRAL, especially during the first few weeks of treatment. If you wish to stand up, you should do so slowly.
- You should monitor your skin and see your doctor in case of any concerns.
- Patients with Parkinson's Disease may have an increased risk of developing melanoma.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how SIMPRAL affects you.
SIMPRAL may cause drowsiness, hallucinations and episodes of sudden onset of sleep in some people. If you experience excessive drowsiness or an episode of sudden onset of sleep (while performing daily activities), do not drive or perform any potentially dangerous activities, and contact your doctor.
Drinking alcohol
Be careful when drinking alcohol while you are taking this medicine. Combining SIMPRAL and alcohol can make you more drowsy or sleepy.
Looking after your medicine
- Keep your tablets in the pack until it is time to take them. If you take the tablets out of the pack they may not keep well.
- Keep your tablets in a cool dry place where the temperature stays below 25°C. Heat and dampness can destroy some medicines.
- A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Store it in a cool dry place away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep it where young children cannot reach it.
Getting rid of any unwanted medicine
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
6. Are there any side effects?
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking SIMPRAL.
SIMPRAL helps most people with Parkinson's disease or RLS, but it may have unwanted side effects in a few people.
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
Do not be alarmed by the following list of side effects. You may not experience any of them.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
Digestive system:
| Speak to your doctor or pharmacist if you have any of these less serious side effects and they worry you. Some of these side effects are more common at the start of treatment and lessen or disappear with time. |
Serious side effects
| Serious side effects | What to do |
Nervous system:
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. These side effects are rare. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What SIMPRAL contains
| Active ingredient (main ingredient) | pramipexole dihydrochloride monohydrate |
| Other ingredients (inactive ingredients) |
|
| Potential allergens |
Do not take this medicine if you are allergic to any of these ingredients.
What SIMPRAL looks like
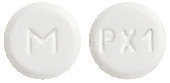
SIMPRAL 0.125 mg is a white to off-white, round, flat-faced tablet, debossed with "PX1" on one side and "M" on the other side (AUST R 173139).
SIMPRAL 0. 25 mg is a white to off-white, biconvex oval shaped tablet, debossed with "PX2" on one side and "M" on one side of breakline on other side (AUST R 173138).
SIMPRAL 1 mg is a white to off-white, round, flat-faced tablet, debossed with "M" over "PX4" on one side and breakline on the other side (AUST R 173137).
Who distributes SIMPRAL
Alphapharm Pty Ltd trading as Viatris
Level 1, 30 The Bond
30-34 Hickson Road
Millers Point NSW 2000
www.viatris.com.au
Phone: 1800 274 276
This leaflet was prepared in December 2023.
SIMPRAL® is a Viatris company trade mark
SIMPRAL_cmi\Dec23/00
Published by MIMS February 2024
 If a further dose increase is necessary the daily dose should be increased by 0.75 mg at weekly intervals up to a maximum dose of 4.5 mg per day.
If a further dose increase is necessary the daily dose should be increased by 0.75 mg at weekly intervals up to a maximum dose of 4.5 mg per day.

 Other events reported by 1% or more of patients treated with pramipexole but reported equally or more frequently in the placebo group were as follows:
Other events reported by 1% or more of patients treated with pramipexole but reported equally or more frequently in the placebo group were as follows: In general, the prevalence of nausea and fatigue was reduced with continued pramipexole therapy.
In general, the prevalence of nausea and fatigue was reduced with continued pramipexole therapy. In patients who had responded to 6-month open label treatment with pramipexole, the administration of placebo led to a rapid decline in their overall conditions and worsening of their RLS symptoms (Figure 1). At the end of the 12-week observation period, 85% of patients treated with placebo had failed treatment, compared to 21% treated with blinded pramipexole; the difference between the treatment groups was highly statistically significant (p < 0.0001).
In patients who had responded to 6-month open label treatment with pramipexole, the administration of placebo led to a rapid decline in their overall conditions and worsening of their RLS symptoms (Figure 1). At the end of the 12-week observation period, 85% of patients treated with placebo had failed treatment, compared to 21% treated with blinded pramipexole; the difference between the treatment groups was highly statistically significant (p < 0.0001).

 Chemical name: (S)-2-amino-4,5,6,7-tetrahydro-6-propylaminobenzothiazole.
Chemical name: (S)-2-amino-4,5,6,7-tetrahydro-6-propylaminobenzothiazole.