SUMMARY CMI
STIVARGA®
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
WARNING: Important safety information is provided in a boxed warning in the full CMI. Read before using this medicine.
1. Why am I using STIVARGA?
STIVARGA contains the active ingredient regorafenib. STIVARGA is used for the treatment of colon, rectal or bowel cancer that has spread to other parts of the body, gastrointestinal stromal tumour (GIST) or liver cancer in patients who have been previously treated with other anticancer medicines.
For more information, see Section 1. Why am I using STIVARGA? in the full CMI.
2. What should I know before I use STIVARGA?
Do not use if you have ever had an allergic reaction to STIVARGA or any of the ingredients listed at the end of the CMI.
Tell your doctor if you have conditions affecting your liver, skin, stomach, bowel or heart, if you have bleeding disorders or taking medicines to thin the blood or if you recently had or are going to have a surgical procedure.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I use STIVARGA? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with STIVARGA and affect how it works.
The most common types of medicines that affect the effectiveness of STIVARGA include antibiotics, antifungals, anti-seizure (antiepileptics), cholesterol lowering medicines, methotrexate and warfarin.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I use STIVARGA?
Swallow whole four STIVARGA tablets with a glass of water at the same time each day after a low-fat meal.
More instructions can be found in Section 4. How do I use STIVARGA? in the full CMI.
5. What should I know while using STIVARGA?
| Things you should do | Remind any doctor, dentist or pharmacist you visit that you are using STIVARGA. Tell your doctor that you are using STIVARGA before any blood tests or surgical procedures. |
| Things you should not do | Do not stop taking your medicine or change the dosage without checking with your doctor. |
| Looking after your medicine | Store it in a cool dry place away from moisture, heat or sunlight where the temperature stays below 25°C. Discard medicine 28 days after opening the bottle |
For more information, see Section 5. What should I know while using STIVARGA? in the full CMI.
6. Are there any side effects?
Less serious side effects include nausea, diarrhoea, headache, fatigue, decreased appetite, dry mouth, tremor, muscle spasm
Serious side effects that require immediate medical attention includes skin reactions (rash, blisters, pain, swelling, itching or peeling of skin), blood in the stools, urine, coughing or vomiting up blood, frequent nose bleeds, signs of liver damage (yellow discoloration of skin and whites of eyes, dark urine, disorientation), severe stomach pain or signs of infection.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI
WARNING: Liver Problems
STIVARGA can cause liver problems which can be serious and in rare cases lead to death. Your doctor will check your liver function before you start taking STIVARGA and monitor your liver function during treatment. Your doctor may need to change your dose or advise you to stop taking STIVARGA. Please see under section SIDE EFFECTS the possible signs of severe liver injury.
FULL CMI
STIVARGA® (Sti-VAR-gah)
Active ingredient(s): [regorafenib]
Consumer Medicine Information (CMI)
This leaflet provides important information about using STIVARGA. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using STIVARGA.
Where to find information in this leaflet:
1. Why am I using STIVARGA?
2. What should I know before I use STIVARGA?
3. What if I am taking other medicines?
4. How do I use STIVARGA?
5. What should I know while using STIVARGA?
6. Are there any side effects?
7. Product details
1. Why am I using STIVARGA?
STIVARGA contains the active ingredient regorafenib. STIVARGA is an antineoplastic (anticancer) agent, it is used to treat cancer by slowing down the growth and spread of cancer cells. It cuts off the blood supply that keeps cancer cells growing
STIVARGA is used to treat:
- colon, rectal or bowel cancer that has spread to other parts of the body in patients who have previously received other treatments,
- gastrointestinal stromal tumour (GIST) in patients who have previously received other treatments
- or liver cancer in patients who have previously been treated with other anticancer medicines.
GIST is a cancer of the stomach and bowel. It is caused by the uncontrolled cells in the wall of the stomach or bowel.
2. What should I know before I use STIVARGA?
Warnings
Do not use STIVARGA if:
- you are allergic to regorafenib, the active ingredient in STIVARGA or any of the ingredients listed at the end of this leaflet.
- you are pregnant or think you might be pregnant
Check with your doctor if you:
- Have liver problems including Gilbert's syndrome.
Treatment with STIVARGA may lead to a higher risk of liver problems and will require blood tests to monitor how your liver is working. - Have or get an infection with signs such as high fever, severe cough, severe sore throat, shortness of breath, burning / pain when urinating, unusual vaginal discharge or irritation, redness, swelling and/or pain in any part of the body. Your doctor may temporarily stop your treatment.
- Have any bleeding problems and you are taking warfarin or another medicine that thins the blood to prevent blood clots. Treatment with STIVARGA may lead to a higher risk of bleeding. Your doctor may conduct blood tests before starting you on STIVARGA
- Have chest pain or any heart problems. Increased heart problems are reported more often in patients over 75 years old. Before starting STIVARGA and during treatment your doctor will check how well your heart is working.
- Have or had a history of high blood pressure and / or aneurysm. STIVARGA can increase your blood pressure. Your doctor will monitor your blood pressure before and during treatment.
- Develop a severe and persistent headache, visual disturbances, seizures or altered mental status.
- Are going to or recently had surgery. STIVARGA might affect the way your wounds heal. Your doctor may stop STIVARGA temporarily before surgery and until your wound has healed.
- Have severe stomach and bowel problems. Your doctor may decide to discontinue treatment.
- Have skin problems. STIVARGA can cause redness, pain, swelling or blisters on the palms of your hands or soles of the feet which may require treatment with creams and shoe cushions. Your doctor may change the dose or stop STIVARGA until your condition improves
Before you use STIVARGA tell your doctor if any of these conditions apply to you. You may need treatment for them, or extra tests may be done.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
- Avoid becoming pregnant while taking STIVARGA, as this may harm your unborn baby. You doctor will advise about using contraception during treatment and for at least eight weeks after treatment if you are of childbearing age.
- Tell your doctor immediately if you are pregnant or intend to become pregnant during treatment with STIGVARGA.
- STIGVARGA should not be used during pregnancy unless necessary. Your doctor will discuss the potential risk of taking STIVARGA during pregnancy.
- STIVARGA may reduce fertility in both men and women. Your doctor will advise you of your options prior to starting treatment.
- Do not breast-feed while taking STIVARGA. Talk to your doctor if you are breastfeeding or intend to breastfeed. This medicine may interfere with the growth and development of your baby
Children and Adolescents
Do not give this medicine to children and adolescents. Safety and effectiveness in children and adolescents have not been established.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and STIVARGA may interfere with each other. These include:
| Antibiotics | Rifampicin, neomycin |
| Antifungals | Ketoconazole, itraconazole, Posaconazole, voriconazole |
| Anti-seizure medicines (Anti-epileptics) | carbamazepine, primidone, phenytoin, phenobarbitone |
| Cholesterol lowering medication | Rosuvastatin, atorvastatin, Fluvastatin |
| Other medications | Warfarin, Methotrexate, St John's Wort |
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect STIVARGA.
4. How do I use STIVARGA?
How much to take
Always take STIVARGA exactly as your doctor has told you to. Check with your doctor or pharmacist if you are not sure
- The recommended daily dose of STIVARGA is four (40 mg tablets) daily. This is equivalent to 160 mg total daily dose.
- Your doctor will usually prescribe STIVARGA for three weeks and then stop for one week. This three week on, one week off treatment period is one cycle of treatment.
- Your doctor may change your dose. Take the dose of STIVARGA that your doctor prescribes for you.
- Follow the instructions provided and use STIVARGA until your doctor tells you to stop.
How to take
- Take four STIVARGA tablets at the same time each day after a low-fat meal (ideally at breakfast).
A low-fat meal contains less than 30% fat. Example of a low-fat meal can include one cup of cereal, 250 mL or one glass of skimmed milk, one slice of toast with jam, apple juice and one cup of coffee or tea.
Swallow the tablets whole with a glass of water.
Do not drink grapefruit juice while taking STIVARGA.
If you forget to use STIVARGA
If you miss a dose, take it as soon as you remember.
Do not take two doses of STIVARGA on the same day to make up for a missed dose from the previous day.
Tell your doctor about any missed dose.
If you use too much STIVARGA
If you think that you have used too much STIVARGA you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
If you take too much STIVARGA it may make the side effects more severe, especially skin reactions (rash, blisters, redness, pain, swelling, itching or peeling of your skin), dysphonia (voice changes or hoarseness), diarrhoea (frequent or loose bowel movements), mucosal inflammation (mouth sores), dry mouth, decreased appetite, hypertension (high blood pressure) and fatigue (excessive tiredness).
5. What should I know while using STIVARGA?
Things you should do
- Tell all doctors, dentists and pharmacists who treat you that you are taking STIVARGA.
- Tell your doctor immediately if you become pregnant during treatment with STIVARGA, or plan to become pregnant.
- Tell your doctor if you are breast-feeding while being treated with STIVARGA.
- If you are about to have any blood tests, tell your doctor that you are taking this medicine. It may interfere with the results of some tests.
- Tell your doctor if you are planning to have surgery or you have a wound that is not healing properly.
Things you should not do
- Do not stop taking your medicine or change the dosage without checking with your doctor
- Do not take STIVARGA to treat any other conditions, unless your doctor tells you to
- Do not give your medicine to anyone else
Driving or using machines
Be careful before you drive or use any machines or tools until you know how STIVARGA affects you.
Drinking alcohol
Tell your doctor if you drink alcohol.
Looking after your medicine
Keep your tablets in the bottle until it is time to take them. If you take the tablets out of the bottle they may not keep well. Do not remove the desiccant from the bottle.
Store it in a cool dry place away from moisture, heat or sunlight where the temperature says below 25°C
Do NOT store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep it where young children cannot reach it.
When to discard your medicine
Once the bottle is opened the medicine is to be discarded after 28 days
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
Gastrointestinal System Related
| Speak to your doctor if you have any of these less serious side effects and they worry you. These are the more common side effects, other side effects not listed may also occur in some patients. Tell your doctor if you notice anything else that is making you feel unwell. |
Some of these side effects can only be found when your doctor does tests from time to time to check your progress.
| These test results include: | What to do |
Blood Test Abnormalities
| Your doctor will tell you if there are any changes in your blood test that might need treatment. |
Serious side effects
| Serious side effects | What to do |
Decreased blood flow to the heart or heart attack
| Call your doctor straight away, or go straight to the >Emergency Department at your nearest hospital if you notice any of these serious side effects. |
| Serious side effects | What to do |
Bleeding from the digestive system
| Call your doctor straight away, or go straight to the Emergency Department at your nearest hospital if you notice any of these serious side effects. |
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What STIVARGA contains
| Active ingredient (main ingredient) | 40 mg regorafenib (as monohydrate) |
| Other ingredients (inactive ingredients) | microcrystalline cellulose croscarmellose sodium magnesium stearate povidone silica colloidal anhydrous iron oxide red iron oxide yellow lecithin macrogol 3350 polyvinyl alcohol purified talc titanium dioxide. |
Do not take this medicine if you are allergic to any of these ingredients.
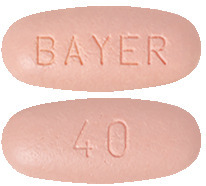
What STIVARGA looks like
STIVARGA is available in 28, 28 (starter pack) or 3 x 28 tablet bottles.
The tablets are light pink, oval embossed with ‘BAYER’ on one side and ‘40’ on the other side.
(AUST R 200553)
Who distributes STIVARGA?
Bayer Australia Ltd
ABN 22 000 138 714
875 Pacific Highway
Pymble NSW 2073
See TGA website (www.ebs.tga.gov.au) for latest Australian Consumer Medicine Information.
® Registered Trademark of Bayer group, Germany.
© Bayer Australia Ltd
All rights reserved.
This leaflet was prepared in 04 July 2023
Published by MIMS September 2023
 For recommended measures and dose modifications in case of worsening of liver function tests considered related to treatment with Stivarga, see Table 2 (see Section 4.4 Special Warnings and Precautions for Use).
For recommended measures and dose modifications in case of worsening of liver function tests considered related to treatment with Stivarga, see Table 2 (see Section 4.4 Special Warnings and Precautions for Use).
 In the pivotal CORRECT study, other adverse reactions observed in patients treated with regorafenib than placebo and that occurred in < 10% (all grades) were: dry skin (8.8% vs 3.2%); proteinuria (8.6% vs 2.4%); alopecia (7.6% vs 1.6%); hypokalaemia (7.6% vs 1.9%); taste disorder (7.6% vs 2.4%); increase in transaminases (7.6% vs 4.4%); hypophosphatemia (6.4% vs 0.8%); increase in lipase (6.2% vs 1.2%); muscle spasms (5.4% vs 2.0%); hypocalcemia (5.8% vs 0.4%); hyponatremia (5.8% vs 2.4%); dry mouth (4.8% vs 2.0%); leukopenia (4.2% vs 0.8%); hypothyroidism (4.2% vs 0.4%); increase in amylase (3.0% vs 0.4%); abnormal international normalised ratio (INR) (2.4% vs 0.8%); hypomagnesemia (2.2% vs 0.4%); tremor (2.0% vs 0%); gastroesophageal reflux (1.4% vs 0%); hyperuricemia (1.2% vs 0%); gastroenteritis (1.2% vs 0.4%); nail disorder (1.0% vs 0%); exfoliative rash (1.4% vs 0.4%); erythema multiforme (0.8% vs 0.4%); gastrointestinal fistula (0.8% vs 0.4%); myocardial ischemia (0.6% vs 0.4%); myocardial infarction (0.4% vs 0%).
In the pivotal CORRECT study, other adverse reactions observed in patients treated with regorafenib than placebo and that occurred in < 10% (all grades) were: dry skin (8.8% vs 3.2%); proteinuria (8.6% vs 2.4%); alopecia (7.6% vs 1.6%); hypokalaemia (7.6% vs 1.9%); taste disorder (7.6% vs 2.4%); increase in transaminases (7.6% vs 4.4%); hypophosphatemia (6.4% vs 0.8%); increase in lipase (6.2% vs 1.2%); muscle spasms (5.4% vs 2.0%); hypocalcemia (5.8% vs 0.4%); hyponatremia (5.8% vs 2.4%); dry mouth (4.8% vs 2.0%); leukopenia (4.2% vs 0.8%); hypothyroidism (4.2% vs 0.4%); increase in amylase (3.0% vs 0.4%); abnormal international normalised ratio (INR) (2.4% vs 0.8%); hypomagnesemia (2.2% vs 0.4%); tremor (2.0% vs 0%); gastroesophageal reflux (1.4% vs 0%); hyperuricemia (1.2% vs 0%); gastroenteritis (1.2% vs 0.4%); nail disorder (1.0% vs 0%); exfoliative rash (1.4% vs 0.4%); erythema multiforme (0.8% vs 0.4%); gastrointestinal fistula (0.8% vs 0.4%); myocardial ischemia (0.6% vs 0.4%); myocardial infarction (0.4% vs 0%).

 In the pivotal GRID study, other adverse reactions observed in less than 10% of Stivarga treated patients and at a higher incidence than in placebo treated patients included the following: hyperbilirubinemia (9.9 vs. 3.0); taste disorders (9.9 vs. 3.0); leukopenia (8.3 vs. 6.1); proteinuria (7.6 vs. 1.5); thrombocytopenia (6.1 vs. 0.0); dry mouth (6.1 vs. 4.6); peripheral sensory neuropathy (6.1 vs. 1.5); hypophosphatemia (5.3 vs. 0.0); dry skin (4.6 vs. 0.0); hypoalbuminaemia (4.5 vs. 0.0); vertigo (4.5 vs. 0.0); hypokalaemia (3.8 vs. 3.0); acute kidney injury (2.3 vs. 0.0); ear discomfort (2.3 vs. 0.0); tinnitus (2.3 vs. 0.0); gastroenteritis (1.5 vs. 0.0); gastrointestinal fistula (1.5 vs. 0.0); hypocalcaemia (1.5 vs. 0.0); increase in lipase (1.5 vs. 0.0); abnormal international normalised ratio (INR) (0.8 vs. 0.0); tremor (0.8 vs. 0.0).
In the pivotal GRID study, other adverse reactions observed in less than 10% of Stivarga treated patients and at a higher incidence than in placebo treated patients included the following: hyperbilirubinemia (9.9 vs. 3.0); taste disorders (9.9 vs. 3.0); leukopenia (8.3 vs. 6.1); proteinuria (7.6 vs. 1.5); thrombocytopenia (6.1 vs. 0.0); dry mouth (6.1 vs. 4.6); peripheral sensory neuropathy (6.1 vs. 1.5); hypophosphatemia (5.3 vs. 0.0); dry skin (4.6 vs. 0.0); hypoalbuminaemia (4.5 vs. 0.0); vertigo (4.5 vs. 0.0); hypokalaemia (3.8 vs. 3.0); acute kidney injury (2.3 vs. 0.0); ear discomfort (2.3 vs. 0.0); tinnitus (2.3 vs. 0.0); gastroenteritis (1.5 vs. 0.0); gastrointestinal fistula (1.5 vs. 0.0); hypocalcaemia (1.5 vs. 0.0); increase in lipase (1.5 vs. 0.0); abnormal international normalised ratio (INR) (0.8 vs. 0.0); tremor (0.8 vs. 0.0). Additional safety data is available from a postregistration phase III trial carried out in Asian patients with CRC (CONCUR). The safety profile in this study was generally consistent with the global phase III CRC trial (CORRECT). Of note, however, a higher incidence of liver enzyme increases was observed in Stivarga treated patients in the CONCUR trial (> 90% East Asian) compared to in the CORRECT trial (~80% Caucasian).
Additional safety data is available from a postregistration phase III trial carried out in Asian patients with CRC (CONCUR). The safety profile in this study was generally consistent with the global phase III CRC trial (CORRECT). Of note, however, a higher incidence of liver enzyme increases was observed in Stivarga treated patients in the CONCUR trial (> 90% East Asian) compared to in the CORRECT trial (~80% Caucasian). Other adverse reactions observed in less than 10% of Stivarga treated patients and at a higher incidence than placebo treated patients included the following: hypophosphatemia (9.9 vs. 2.1), leukopenia (8.8 vs. 3.6), proteinuria (8.8 vs. 1.6), rash (8.6 vs. 8.3), stomatitis (8.3 vs. 2.1), increase in lipase (7.2 vs. 3.1), alopecia (7.0 vs. 2.6), hypokalemia (7.0 vs. 2.6), hypothyroidism (6.7 vs. 0.0), headache (6.4 vs. 6.2), hyponatremia (5.9 vs. 3.1), dry mouth (5.9 vs. 4.7), hypomagnesaemia (3.2 vs. 0.0), taste disorder (3.2 vs. 1.0), increase in amylase (2.9 vs. 0.0), mucosal inflammation (2.9 vs. 0.0), hypocalcemia (2.4 vs. 0.0), pancreatitis (1.6 vs. 0.0), exfoliative rash (1.3 vs. 0.0), tremor (1.3 vs. 0.0), hyperuricaemia (1.1 vs. 1.0), erythema multiforme (0.8 vs. 0.0), myocardial ischemia (0.8 vs. 0.0), gastroenteritis (0.5 vs. 0.0), gastrointestinal fistula (0.3 vs. 0.0), myocardial infarction (0.3 vs. 0.0), nail disorder (0.3 vs. 0.0).
Other adverse reactions observed in less than 10% of Stivarga treated patients and at a higher incidence than placebo treated patients included the following: hypophosphatemia (9.9 vs. 2.1), leukopenia (8.8 vs. 3.6), proteinuria (8.8 vs. 1.6), rash (8.6 vs. 8.3), stomatitis (8.3 vs. 2.1), increase in lipase (7.2 vs. 3.1), alopecia (7.0 vs. 2.6), hypokalemia (7.0 vs. 2.6), hypothyroidism (6.7 vs. 0.0), headache (6.4 vs. 6.2), hyponatremia (5.9 vs. 3.1), dry mouth (5.9 vs. 4.7), hypomagnesaemia (3.2 vs. 0.0), taste disorder (3.2 vs. 1.0), increase in amylase (2.9 vs. 0.0), mucosal inflammation (2.9 vs. 0.0), hypocalcemia (2.4 vs. 0.0), pancreatitis (1.6 vs. 0.0), exfoliative rash (1.3 vs. 0.0), tremor (1.3 vs. 0.0), hyperuricaemia (1.1 vs. 1.0), erythema multiforme (0.8 vs. 0.0), myocardial ischemia (0.8 vs. 0.0), gastroenteritis (0.5 vs. 0.0), gastrointestinal fistula (0.3 vs. 0.0), myocardial infarction (0.3 vs. 0.0), nail disorder (0.3 vs. 0.0).
 The addition of Stivarga to BSC resulted in significantly longer survival compared to placebo plus BSC (p = 0.005178 stratified log rank test) with a hazard ratio of 0.774 [95% CI 0.636, 0.942] and a median OS of 6.4 months vs. 5.0 months (see Table 11 and Figure 1). PFS was significantly longer in patients receiving Stivarga plus BSC (HR: 0.494, p < 0.000001, see Table 11 and Figure 2).
The addition of Stivarga to BSC resulted in significantly longer survival compared to placebo plus BSC (p = 0.005178 stratified log rank test) with a hazard ratio of 0.774 [95% CI 0.636, 0.942] and a median OS of 6.4 months vs. 5.0 months (see Table 11 and Figure 1). PFS was significantly longer in patients receiving Stivarga plus BSC (HR: 0.494, p < 0.000001, see Table 11 and Figure 2).

 The response rate (complete response or partial response) was 1% and 0.4% for Stivarga and placebo treated patients respectively, however the difference was not statistically significant (p = 0.188432, 1 sided). The disease control rate (complete response or partial response or stable disease) was significantly higher in patients treated with Stivarga (41.0% vs 14.9%, p < 0.000001, 1 sided).
The response rate (complete response or partial response) was 1% and 0.4% for Stivarga and placebo treated patients respectively, however the difference was not statistically significant (p = 0.188432, 1 sided). The disease control rate (complete response or partial response or stable disease) was significantly higher in patients treated with Stivarga (41.0% vs 14.9%, p < 0.000001, 1 sided).

 At progression, crossover from placebo to Stivarga was allowed. Fifty six patients randomised to placebo (85%) crossed over to Stivarga and 41 patients randomised to Stivarga (31%) continued on Stivarga. The median secondary PFS (as measured by the investigator) was 5.0 months for those randomised to placebo and 4.5 months for those randomised to Stivarga.
At progression, crossover from placebo to Stivarga was allowed. Fifty six patients randomised to placebo (85%) crossed over to Stivarga and 41 patients randomised to Stivarga (31%) continued on Stivarga. The median secondary PFS (as measured by the investigator) was 5.0 months for those randomised to placebo and 4.5 months for those randomised to Stivarga. The median duration of treatment was 3.6 months on Stivarga and 1.9 months on placebo.
The median duration of treatment was 3.6 months on Stivarga and 1.9 months on placebo.



