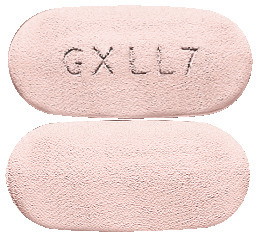
SUMMARY CMI
TELZIR
Consumer Medicine Information (CMI) summary
The full CMI on the next page has more details. If you are worried about using this medicine, speak to your doctor or pharmacist.
1. Why am I taking TELZIR?
TELZIR contains the active ingredient fosamprenavir which belongs to a group of antiretroviral medicines called protease inhibitors. TELZIR is taken with ritonavir and is used to slow down the progression of human immunodeficiency virus (HIV) infection which can lead to Acquired Immune Deficiency Syndrome (AIDS) and other related illnesses (e.g. AIDS-related Complex (ARC)).
For more information, see Section 1. Why am I taking TELZIR? in the full CMI.
2. What should I know before I use TELZIR?
Do not use if you have ever had an allergic or hypersensitivity reaction to fosamprenavir, amprenavir, ritonavir or any of the ingredients listed at the end of the CMI.
Talk to your doctor if you have any other medical conditions, take any other medicines, or are pregnant or plan to become pregnant or are breastfeeding.
For more information, see Section 2. What should I know before I take TELZIR? in the full CMI.
3. What if I am taking other medicines?
Some medicines may interfere with TELZIR and affect how it works.
A list of these medicines is in Section 3. What if I am taking other medicines? in the full CMI.
4. How do I take TELZIR?
- Adults over the age of 18 in treatment-naïve patients: the dose is 1400 mg (two tablets) once a day with 200 mg ritonavir once a day or 700 mg (one tablet) twice a day with 100 mg ritonavir twice a day.
- Adults over the age of 18 in protease inhibitor experienced patients: the dose is 700 mg (one tablet) with 100 mg ritonavir twice a day in combination with other antiretroviral agents.
- In children and adolescents between 6 and 18: 700 mg (one tablet) twice a day plus 100 mg ritonavir twice a day.
More instructions can be found in Section 4. How do I take TELZIR? in the full CMI.
5. What should I know while using TELZIR?
| Things you should do |
|
| Things you should not do |
|
| Driving or using machines |
|
| Looking after your medicine |
|
For more information, see Section 5. What should I know while using TELZIR? in the full CMI.
6. Are there any side effects?
Side effects that have been reported include: nausea, vomiting, diarrhoea, flatulence, abdominal pain, headache, rash, fatigue, feeling tired, an altered sensation in your mouth, dizziness, muscle pain, tenderness and weakness.
For more information, including what to do if you have any side effects, see Section 6. Are there any side effects? in the full CMI.
FULL CMI
TELZIR
Active ingredient(s): fosamprenavir
Consumer Medicine Information (CMI)
This leaflet provides important information about using TELZIR. You should also speak to your doctor or pharmacist if you would like further information or if you have any concerns or questions about using TELZIR.
Where to find information in this leaflet:
1. Why am I taking TELZIR?
2. What should I know before I take TELZIR?
3. What if I am taking other medicines?
4. How do I take TELZIR?
5. What should I know while taking TELZIR?
6. Are there any side effects?
7. Product details
1. Why am I taking TELZIR?
TELZIR contains the active ingredient fosamprenavir. TELZIR belongs to a group of antiretroviral medicines called protease inhibitors.
TELZIR is taken with another HIV medicine called ritonavir and is used to slow down the progression of human immunodeficiency virus (HIV) infection which can lead to Acquired Immune Deficiency Syndrome (AIDS) and other related illnesses (e.g. AIDS-related Complex (ARC)).
The low doses of ritonavir you take together with TELZIR are designed to increase the amount of the active ingredient, amprenavir, available to control your HIV infection.
TELZIR does not cure AIDS or HIV infection however it slows down production of HIV in the body. In this way it stops ongoing damage to the body's immune system which fights infection.
You can still pass on HIV when taking this medicine through sexual activity or through passing on blood or bodily secretions which carry the HIV virus, although the risk is lowered by taking antiretroviral therapy.
You should use proper precautions to prevent this from occurring. Discuss with your doctor the precautions needed to avoid infecting other people.
While taking TELZIR and/or any other therapy for HIV, you may continue to develop other infections and other complications of HIV infection. You should keep in regular contact with your doctor.
It is important to read the patient information relating to ritonavir before you start taking this medicine. Ask your doctor or pharmacist for a copy.
2. What should I know before I take TELZIR?
Warnings
Do not use TELZIR if:
- you are allergic or had a hypersensitivity reaction to fosamprenavir, amprenavir, or any of the ingredients listed at the end of this leaflet.
Always check the ingredients to make sure you can use this medicine.
- you are allergic to ritonavir or any of the ingredients in that medicine
- you are pregnant, trying to become pregnant or are breastfeeding
- you are taking any of the following medicines: astemizole, terfenadine, rifampicin, telaprevir, boceprevir, simeprevir, paritaprevir, midazolam, triazolam, sildenafil, dihydroergotamine, ergotamine, cisapride, pimozide, quetiapine, lurasidone, flecainide, propafenone, St John's Wort
Check with your doctor if you:
- have any other medical conditions
- are allergic to food, dyes, preservatives or any other medicines (eg sulfonamide)
- have diabetes mellitus or haemophilia
- have or have ever had any liver problems, for example jaundice, hepatitis, a virus affecting your liver, an enlarged liver or liver scarring (cirrhosis)
If you have liver disease you will need to have your dose of TELZIR and/or ritonavir adjusted. It is not possible to reduce the dose of TELZIR tablets to less than 700 mg, so people with moderate or severe liver disease must not be treated with TELZIR tablets. If you are unsure about your level of liver disease consult your doctor who may carry out additional testing to see whether the TELZIR/ritonavir combination is suitable for you.
- take any medicines for any other condition
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy and breastfeeding
Talk to your doctor if you are pregnant or intend to become pregnant.
Talk to your doctor if you are breastfeeding or intend to breastfeed.
Your doctor will discuss with you the risks and benefits involved of using TELZIR with ritonavir whilst pregnant or breastfeeding.
If you are taking the contraceptive pill, it is recommended that you use an alternative method of contraception (e.g. a condom) to prevent pregnancy while you are taking TELZIR.
Symptoms of infection and inflammation
People with advanced HIV infection (AIDS) have weak immune systems and are more likely to develop serious infections (opportunistic infections). When they start treatment, the immune system becomes stronger and so the body starts to fight infections.
Symptoms of infection and inflammation may develop, caused by either:
- old, hidden infections flaring up at the body fights them
- the immune system attacking healthy body tissue (autoimmune disorders)
The symptoms of autoimmune disorders may develop many months after you start taking medications to treat your HIV infection.
Symptoms may include:
- muscle weakness and/or muscle pain
- joint pain or swelling
- weakness beginning in the hands or feet and moving towards the trunk of the body
- palpitations or tremor
- hyperactivity (excessive restlessness and movement)
If you get symptoms of infection or if you notice any of the symptoms above, tell your doctor immediately. Do not take other medicines for the infection without your doctor's advice.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other medicines, including any medicines, vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
The following medicines can interact with TELZIR and ritonavir and must not be taken with TELZIR and ritonavir:
- astemizole and terfenadine, which are used to treat hayfever
- rifampicin, used to treat tuberculosis
- telaprevir, boceprevir, simeprevir, paritaprevir and similar medicines used to treat hepatitis C
- nevirapine, a medicine used to treat HIV
- midazolam and triazolam, medicines which induce sedation
- sildenafil when it is used to treat increased pressure in the blood vessels to your lungs
- dihydroergotamine and ergotamine which are used to treat migraine
- cisapride which is used to treat gastric reflux
- pimozide which is used to treat chronic psychotic disorders
- alfuzosin, a medicine used in men to treat benign prostatic hyperplasia
- quetiapine, a medicine used to treat schizophrenia, bipolar disorder and major depressive disorder
- lurasidone, a medicine used to treat schizophrenia and bipolar disorder
- flecainide which is used to treat heart conditions
- propafenone which is used to treat heart conditions
- herbal treatments such as St Johns Wort
TELZIR and ritonavir may interact with certain other medications. The use of these medicines, together with the TELZIR/ritonavir combination, should only take place on the basis of medical advice which may include monitoring decreased medicine effects, monitoring your blood and decreasing dosages:
- antibiotics such as rifabutin, clarithromycin, dapsone and erythromycin
- antifungals including ketoconazole, itraconazole
- halofantrine, an antimalarial medicine
- benzodiazepines such as alprazolam and clorazepam
- calcium channel blockers such as diltiazem, nicardipine, nifedipine, nimodipine, felodipine, verapamil, amlodipine, and isradipine
- medicines used to lower your cholesterol including atorvastatin, lovastatin and simvastatin
- erectile dysfunction medicines including sildenafil, tadalafil, vardenafil
- non-nucleoside reverse transcriptase inhibitors such as efavirenz, nevirapine and delavirdine
- opioids, for example methadone
- steroids such as oestrogens, progestogens and some glucocorticoids
- inhaled/nasal steroid including fluticasone
- immunosuppressants and medicines used to suppress your immune system such as ciclosporin, tacrolimus
- clozapine, an antipsychotic medicine
- carbamazepine, a medicine used to in seizures
- famotidine, nizatidine, ranitidine, cimetidine, medicines used to decrease stomach acid production
- paroxetine, a medicine used to treat depression
- loratadine, a medicine used to treat hayfever
- maraviroc and dolutegravir, medicines used to treat HIV
- medications used to treat several types of cancers such as dasatinib, nilotinib, ibrutinib, vinblastine and everolimus
If you are taking certain medicines that can cause serious side effects, such as amiodarone, phenobarbitone, phenytoin, lidocaine, tricyclic antidepressants, quinidine and warfarin, at the same time as you are taking the TELZIR/ritonavir combination, your doctor may carry out additional blood tests to minimise any potential safety problems.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect TELZIR.
4. How do I take TELZIR?
How much to take
Adults over the age of 18:
- Treatment-naïve patients: the dose of TELZIR is 1400 mg (two tablets) once a day with 200 mg ritonavir once a day or 700 mg (one tablet) twice a day with 100 mg ritonavir twice a day
- Protease inhibitor experienced patients: the recommended dose is 700 mg (one tablet) with 100 mg ritonavir twice a day in combination with other antiretroviral agents. The once daily administration of TELZIR plus ritonavir is not recommended in protease inhibitor experienced patients.
Children and adolescents between 6 and 18 years of age:
- If the child weighs over 39 kg and can swallow the tablets whole then the adult dosing regimen of 700 mg (one tablet) twice a day plus 100 mg ritonavir twice a day can be used. Ritonavir 100 mg capsules can be used by children and adolescents who weigh more than 33 kg and can swallow the capsules whole.
The use of TELZIR in combination with ritonavir has not been established in children less than 6 years of age.
Follow the instructions provided and use TELZIR until your doctor tells you to stop.
How to take TELZIR
- Swallow the tablets whole with a glass of water.
- You can take TELZIR with or without food.
If you forget to take TELZIR
TELZIR should be used regularly at the same time each day.
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to.
Do not take a double dose to make up for the dose you missed.
If you take too much TELZIR
If you think that you have used too much TELZIR, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre
(by calling 13 11 26), or - contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
5. What should I know while taking TELZIR?
Things you should do
Tell your doctor straight away if you:
- become pregnant or intend to become pregnant
- have not taken TELZIR as intended
Remind any doctor, dentist or pharmacist you visit that you are taking TELZIR.
Stay in regular contact with your doctor
TELZIR helps to control your condition, but it is not a cure for HIV infection. You need to keep taking it every day to stop your illness from getting worse. Because TELZIR does not cure HIV infections, you may still develop other infections and illnesses linked to HIV.
Things you should not do
- Do not stop taking this medicine suddenly or change the dose.
- Do not give this medicine to anyone else, even if their symptoms seem similar to yours.
- Do not use this medicine to treat any other complaints unless your doctor tells you to.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how TELZIR affects you.
Looking after your medicine
Follow the instructions on the pack on how to take care of your medicine properly.
Store it in a cool dry place (below 30°C) away from moisture, heat or sunlight; for example, do not store it:
- in the bathroom or near a sink, or
- in the car or on window sills.
Keep it where young children cannot reach it.
Getting rid of any unwanted medicine
If you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
Do not use this medicine after the expiry date.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
Within the first few weeks of treatment with anti-HIV medicines, some people, particularly those that have been HIV positive for some time, may develop inflammatory reactions (e.g. pain, redness, swelling, high temperature) which may resemble an infection and may be severe. It is thought that these reactions are caused by a recovery in the body's ability to fight infections, previously suppressed by HIV.
If you become concerned about any new symptoms, or any changes in your health after starting HIV treatment, discuss with your doctor immediately.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
Less serious side effects
| Less serious side effects | What to do |
Gastrointestinal
Musculoskeletal:
| Speak to your doctor if you have any of these less serious side effects and they worry you. |
In some individuals, treatment with a combination of antiretroviral medicines that includes a protease inhibitor may find their body shape changes due to an increase in body fat. It is not yet known what causes these changes, or whether they have any long-term effects on your health. If you notice changes in your body shape, speak to your doctor.
Your doctor will ask you to undertake blood tests regularly to monitor for any abnormalities (blood lipids and blood sugar). Increases in liver enzymes and blood fats have been reported in patients taking TELZIR and ritonavir.
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell.
Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What TELZIR contains
| Active ingredient (main ingredient) | Fosamprenavir as fosamprenavir sodium |
| Other ingredients (inactive ingredients) | croscarmellose sodium colloidal silicon dioxide magnesium stearate microcrystalline cellulose povidone K30 The tablet coating contains: glycerol triacetate hypromellose iron oxide red (E172) titanium dioxide (E171) |
Do not take this medicine if you are allergic to any of these ingredients.
What TELZIR looks like
TELZIR tablets are pink, film-coated, capsule-shaped, biconvex tablets, debossed GX LL7 on one face (AUST R 101604). They are available in bottles of 60 tablets.
Who distributes TELZIR
ViiV Healthcare Pty Ltd
Level 4, 436 Johnson Street
Abbotsford VIC 3067
Australia
Licenced from Vertex Pharmaceuticals Inc., Cambridge MA 02139 USA.
Trademarks are owned by or licenced to the ViiV Healthcare group of companies.
© 2022 ViiV Healthcare group of companies or its licensor.
This leaflet was prepared in November 2022.
Version 13.0
Published by MIMS March 2023






 In antiretroviral naïve patients (APV30002) receiving Telzir/ritonavir in combination with abacavir and lamivudine, drug hypersensitivity was commonly# reported. All cases were reported as possibly related to abacavir. In cases of reported drug hypersensitivity, abacavir was discontinued and an alternative antiretroviral drug substituted. Few patients withdrew from the study due to these events. (#: Common defined as ≥ 1%, < 10%.)
In antiretroviral naïve patients (APV30002) receiving Telzir/ritonavir in combination with abacavir and lamivudine, drug hypersensitivity was commonly# reported. All cases were reported as possibly related to abacavir. In cases of reported drug hypersensitivity, abacavir was discontinued and an alternative antiretroviral drug substituted. Few patients withdrew from the study due to these events. (#: Common defined as ≥ 1%, < 10%.) Routine pharmacovigilance identified oral paraesthesia with fosamprenavir as a safety signal. A review of the key clinical trials for fosamprenavir revealed a trend towards oral paraesthesia (and related AEs), with a frequency category of common (> 1% - < 10%).
Routine pharmacovigilance identified oral paraesthesia with fosamprenavir as a safety signal. A review of the key clinical trials for fosamprenavir revealed a trend towards oral paraesthesia (and related AEs), with a frequency category of common (> 1% - < 10%). The virologic response based upon baseline phenotype was assessed. Baseline isolates from PI experienced patients responding to fosamprenavir/ritonavir twice daily had a median shift in susceptibility to amprenavir relative to a standard wild type reference strain of 0.7 (range: 0.1 to 5.4, n = 62), and baseline isolates from individuals failing therapy had a median shift in susceptibility of 1.9 (range: 0.2 to 14, n = 29). Because this was a select patient population, these data do not constitute definitive clinical susceptibility breakpoints. Additional data are needed to determine clinically relevant breakpoints for Telzir.
The virologic response based upon baseline phenotype was assessed. Baseline isolates from PI experienced patients responding to fosamprenavir/ritonavir twice daily had a median shift in susceptibility to amprenavir relative to a standard wild type reference strain of 0.7 (range: 0.1 to 5.4, n = 62), and baseline isolates from individuals failing therapy had a median shift in susceptibility of 1.9 (range: 0.2 to 14, n = 29). Because this was a select patient population, these data do not constitute definitive clinical susceptibility breakpoints. Additional data are needed to determine clinically relevant breakpoints for Telzir. The median baseline CD4 count was low in both groups. A total of 55% of subjects had a CD4+ cell count < 200 cells/mm3 at baseline, and 20% of subjects had CD4+ cell counts < 50 cells/mm3. Overall, the baseline characteristics demonstrate that a more advanced treatment naive population was enrolled in this study compared to the population of previous studies in treatment naive subjects.
The median baseline CD4 count was low in both groups. A total of 55% of subjects had a CD4+ cell count < 200 cells/mm3 at baseline, and 20% of subjects had CD4+ cell counts < 50 cells/mm3. Overall, the baseline characteristics demonstrate that a more advanced treatment naive population was enrolled in this study compared to the population of previous studies in treatment naive subjects. The results from study APV 30002 indicates that, in antiretroviral naive patients, fosamprenavir given once daily in combination with low dose ritonavir as part of a regimen including abacavir and lamivudine (given twice daily) showed comparable efficacy over 48 weeks compared to nelfinavir given twice daily in combination with abacavir/lamivudine.
The results from study APV 30002 indicates that, in antiretroviral naive patients, fosamprenavir given once daily in combination with low dose ritonavir as part of a regimen including abacavir and lamivudine (given twice daily) showed comparable efficacy over 48 weeks compared to nelfinavir given twice daily in combination with abacavir/lamivudine. Mean plasma HIV-1 RNA Average Area Under the Curve Minus Baseline (AAUCMB) values at week 48 are summarised in Table 13 for the ITT population observed analysis.
Mean plasma HIV-1 RNA Average Area Under the Curve Minus Baseline (AAUCMB) values at week 48 are summarised in Table 13 for the ITT population observed analysis. Table 14 provides plasma HIV-1 RNA at week 48 for < 400 and < 50 copies/mL using the ITT rebound or discontinued = failure (RD = F) analysis.
Table 14 provides plasma HIV-1 RNA at week 48 for < 400 and < 50 copies/mL using the ITT rebound or discontinued = failure (RD = F) analysis. The fosamprenavir/ritonavir twice daily regimen and the lopinavir/ritonavir twice daily regimen showed similar immunological improvements through 48 weeks of treatment, as measured by median CD4+ cell count and change from baseline.
The fosamprenavir/ritonavir twice daily regimen and the lopinavir/ritonavir twice daily regimen showed similar immunological improvements through 48 weeks of treatment, as measured by median CD4+ cell count and change from baseline. Administration of the fosamprenavir oral tablet formulation (1400 mg) with a high fat meal did not alter plasma amprenavir pharmacokinetics as compared to the administration of this formulation in the fasted state. Fosamprenavir tablets may be taken without regard to food intake.
Administration of the fosamprenavir oral tablet formulation (1400 mg) with a high fat meal did not alter plasma amprenavir pharmacokinetics as compared to the administration of this formulation in the fasted state. Fosamprenavir tablets may be taken without regard to food intake. Currently there are insufficient pharmacokinetic data in children < 2 years of age to support dosing in this age group.
Currently there are insufficient pharmacokinetic data in children < 2 years of age to support dosing in this age group.
