
What is in this leaflet?
Please read this leaflet carefully before you start 3TC Tablets.
This leaflet answers some common questions about 3TC tablets. It does not contain all of the available information.
It does not take the place of talking to your doctor or pharmacist (also known as a chemist).
All medicines have benefits and risks. Sometimes new risks are found even when a medicine has been used for many years. Your doctor has weighed the risks of you taking 3TC against the expected benefits it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the tablets. You may need to read it again.
What 3TC tablets are used for
3TC tablets contain lamivudine which belongs to a group of medicines called antivirals.
3TC is used together with other antivirals to slow down the progression of human immunodeficiency virus (HIV) infection, which can lead to Acquired Immune Deficiency Syndrome (AIDS) and other related illnesses (eg AIDS-related Complex or ARC).
3TC does not cure AIDS or kill the HIV virus, but prevents further damage to the immune system by stopping production of new viruses.
You can still pass on HIV when taking this medicine, although the risk is lowered by effective antiretroviral therapy. Discuss with your doctor the precautions needed to avoid infecting other people.
You will still be able to pass on the HIV virus by sexual activity or by contamination with infected blood. You should still use proper precautions.
While taking 3TC and/or any other therapy for HIV disease, you may continue to develop other infections and other complications of HIV infection. You should keep in regular contact with the doctor who is looking after you.
3TC tablets are not addictive.
Your doctor may have prescribed 3TC tablets for another reason.
Ask your doctor if you have any questions about why 3TC tablets have been prescribed for you.
Before you take 3TC tablets
When you must not take them
- Do not take 3TC tablets if you have ever had an allergic reaction to lamivudine or any of the ingredients listed at the end of this leaflet.
Read the Side Effects section to find out symptoms of allergy. - Do not take 3TC tablets if you are pregnant, trying to become pregnant or breastfeeding, unless your doctor says you should.
Your doctor will discuss the risks and benefits of using 3TC tablets if you are pregnant or breastfeeding. - Do not use 3TC tablets to treat any other conditions unless your doctor says that you should do so.
- Do not give 3TC tablets to anyone else, even though their symptoms may sound similar to yours.
- Do not take 3TC tablets after the expiry or "use by" date (EXP) printed on the pack.
If you take it after the expiry date has passed, it may not work as well. - Do not take 3TC tablets if the packaging is torn or shows signs of tampering.
If you're not sure whether you should be taking 3TC tablets, talk to your doctor.
Before you start to take them
You must tell your doctor if:
- you are allergic to foods, dyes, preservatives or any other medicines.
- you have, or have ever had, liver problems, for example jaundice, hepatitis, virus affecting the liver, enlarged liver or liver scarring (cirrhosis) or if you have any risk factors for liver problems, e.g. excessive alcohol intake, illegal intravenous drug use with shared equipment, iron or copper storage disorders
- you have, or have ever had, kidney problems.
- you have, or have ever had, problems with your pancreas.
- you have diabetes.
- you have any other illness, including those that you think are not related to HIV infection.
When you stop taking 3TC Tablets
If you have a long-standing viral infection of your liver (hepatitis B) it may flare up. This can cause serious illness particularly if your liver is already not working very well. If you have both HIV and hepatitis B, when you stop taking your 3TC tablets, your doctor is likely to arrange tests from time to time to check how well your liver is working and to measure virus levels.
Taking other medicines
Tell your doctor if you are taking any other medicines, including medicines you buy without a prescription from a pharmacy, supermarket or health food shop.
Some medicines may affect the way others work. Your doctor or pharmacist will be able to tell you what to do when taking 3TC tablets with other medicines.
How to take 3TC tablets
Your doctor will tell you how many 3TC tablets to take and how often to take them. You will also find this information on the label of your medicine.
Do not take extra tablets. Do not take the tablets more often than you have been told.
How much to take
Adults and adolescents 12 years and older the usual dose is one 150 mg tablet twice a day or 300 mg once a day. Your doctor may prescribe a different dosage.
For younger children between 3 months to 12 years of age the dose of 3TC will depend on their weight in kilograms (kg). If you are giving 3TC tablets to a child, follow your doctor's instructions.
3TC oral solution is also available for younger patients, or those who cannot swallow tablets.
How to take them
3TC tablets should be swallowed whole with a glass of water.
If you cannot swallow the tablet(s), you may crush and combine them with a small amount of food or drink, and take all the dose immediately.
How long to take them
Because your medicine helps to control your condition, but does not cure it, you will need to take the tablets every day. Do not stop taking your medicine without first talking to your doctor.
If you forget to take them
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to. Otherwise, take it as soon as you remember, then go back to taking it as you would normally.
Do not take a double dose to make up for the dose that you missed.
If you take too much (overdose)
Immediately telephone your doctor or Poisons Information Centre (telephone 131126), or in New Zealand the Poisons Centre number is 0800 POISON or 0800 764 766, or go to accident and emergency at your nearest hospital, if you think you or anyone else may have taken too many 3TC tablets. Do this even if there are no signs of discomfort or poisoning. This may need urgent medical attention.
Keep telephone numbers for these places handy.
If you are not sure what to do, contact your doctor or pharmacist.
While you are taking 3TC tablets
Things you must do
Tell your doctor or pharmacist that you are taking 3TC tablets if you are about to be started on any other medicines.
Tell your doctor if you become pregnant or are trying to become pregnant.
Tell your doctor if, for any reason, you have not taken your medicine exactly as prescribed. Otherwise, your doctor may think that it was not effective and change your treatment unnecessarily.
Things you must not do
Do not stop taking 3TC tablets, or change the dose without first checking with your doctor.
Do not give this medicine to anyone else, even if their symptoms seem similar to yours.
Do not use 3TC tablets to treat any other complaints unless your doctor says to.
Things to be careful of
Be careful driving or operating machinery until you know how 3TC tablets affect you.
Side-Effects
Check with your doctor as soon as possible if you have any problems while taking 3TC tablets, even if you do not think the problems are connected with the medicine or are not listed in this leaflet.
Like all medicines, 3TC tablets can cause some side-effects. If they occur, they are most likely to be minor and temporary. However, some may be serious and need medical attention.
Tell your doctor or pharmacist if you notice any of the following and they worry you.
The most common side-effects (could affect at least one to ten in every 100 people) are:
- nausea, vomiting
- diarrhoea
- upper abdominal pain
- headache
- high temperature
- lethargy, fatigue
- hair loss
- joint and muscle pain
- skin rash (without any other illness).
- increased bruising or bleeding
Uncommon side-effects (could affect less than one in every 100 people) are:
- increases in enzymes produced by the liver
- anaemia (low red blood cell count)
- neutropenia (low white blood cell count)
- reduction in the number of platelets (blood cells important for blood clotting).
If the production of red blood cells is reduced, you may have symptoms of tiredness or breathlessness.
A reduction in your white blood cell count can make you more prone to infection. If you have a low platelet count, you may notice that you bruise more easily.
Rare side-effects (could affect less than one in every 1,000 people) are:
- breakdown of muscle tissue
- increases of an enzyme called amylase
- inflammation of the pancreas (pancreatitis)
- lactic acidosis
Very rare side-effects (could affect less than one in every 10,000 people) are:
- unusual feelings in any part of the body, such as numbness, burning, tingling or pins and needles
- numbness or weakness of the arms and legs
- severe anaemia
Fat loss or fat gain has been observed with combined antiretroviral therapy. A causal relationship for this has not been established. Should any change in body shape be noticed, seek medical advice.
Other effects may show up in blood tests including increased blood levels of sugar, fatty acids (triglycerides) and cholesterol.
Within the first few weeks of treatment with anti-HIV medicines, some people, particularly those that have been HIV positive for some time, may develop inflammatory reactions (eg pain, redness, swelling, high temperature) which may resemble an infection and may be severe. It is thought that these reactions are caused by a recovery in the body's ability to fight infections, previously suppressed by HIV. If you become concerned about any new symptoms, or any changes in your health after starting HIV treatment, please discuss with your doctor immediately.
Ask your doctor or pharmacist to answer any questions you may have.
Some people are allergic to medicines.
If you have any of the following symptoms soon after starting to take your medicine, DO NOT TAKE ANY MORE 3TC TABLETS and tell your doctor IMMEDIATELY or go to the accident and emergency department at your nearest hospital:
- Skin troubles such as lumpy skin rash or "hives".
- Swelling of the face, lips, mouth or throat which may cause difficulty in swallowing or breathing.
- Wheezing, chest pain or tightness.
- Fainting.
These are very serious side effects. If you have them, you may have a serious allergic reaction. You may need urgent medical attention or hospitalisation. All these side effects are very rare.
If you have any of the following symptoms soon after starting to take your medicine, DO NOT TAKE ANY MORE 3TC TABLETS and tell your doctor IMMEDIATELY or go to the accident and emergency department at your nearest hospital:
- Severe stomach pain or cramps.
- Nausea.
- Vomiting.
These side effects may be due to a condition called pancreatitis.
If you are on medication for HIV and become very sick, with fast breathing, stop taking 3TC tablets and consult your doctor immediately. You may have a condition known as "lactic acidosis". The fast breathing is due to high acid levels in the blood. Your liver may not be working properly and gets big and fatty. This can be life threatening. This illness occurs more often in women than men.
See your doctor if you feel generally unwell with loss of appetite, nausea, vomiting, itching, yellowness of the skin or eyes or dark coloured urine, or if the blood tests of your liver function are abnormal. It is likely you will have to stop taking 3TC tablets.
Tell your doctor if you notice anything else that is making you feel unwell, even if it is not on this list. Some people may get other side effects while taking 3TC tablets. If you are concerned, talk to your doctor or pharmacist.
Ask your doctor or pharmacist if you don't understand anything in this list.
Do not be alarmed by this list of possible side-effects. You may not experience any of them.
After taking 3TC tablets
Storage
Keep the tablets where children cannot reach them. A locked cupboard at least one-and-a half metres above the ground is a good place to store them.
Keep 3TC tablets in a cool, dry place where it stays below 30°C.
Do not store the tablets, or any other medicine, in a bathroom or near a sink.
Do not leave them in the car or on window sills. Heat and dampness can destroy some medicines.
Keep your 3TC tablets in the bottle with the cap tightly closed until you take them. If you take 3TC tablets out of their pack they may not keep well.
Disposal
If your doctor tells you to stop taking 3TC tablets, or the tablets have passed their expiry date, ask your pharmacist what to do with any tablets left over.
Product description
What 3TC tablets look like.
3TC 150 mg Tablets are white, film-coated, modified diamond shaped tablets engraved "GX CJ7" on one face.
3TC 300 mg Tablets are grey, film-coated, modified diamond shaped tablets engraved "GX EJ7" on one face.
3TC tablets are supplied in a white high-density polyethylene bottle, with a plastic cap. Each bottle contains 30 (300 mg) or 60 (150 mg) tablets and is in a carton.
Ingredients
3TC tablets contain either 150 mg or 300 mg of lamivudine per tablet.
3TC Tablets also contain Microcrystalline cellulose (460), magnesium stearate (572), titanium dioxide (171), hypromellose (464), sodium starch glycollate, macrogol 400 and polysorbate 80 (433) and purified water. The 300 mg tablets also contain black iron oxide (E172).
Sponsor
ViiV Healthcare Pty Ltd
Level 4, 436 Johnston Street
Abbotsford, Victoria, 3067
Australia.
Further Information
This is not all the information that is available on 3TC tablets. If you have any more questions or are not sure about anything, ask your doctor or pharmacist.
Pharmaceutical companies are not in a position to give people an individual diagnosis or medical advice. Your doctor or pharmacist is the best person to give you the individual advice you need. You may also be able to find out more information about your disease from books, for example in public libraries.
Counselling is also available from your local AIDS council.
Do not throw this leaflet away. You may need to read it again.
This leaflet was prepared on
3 February 2021
The information provided applies only to: 3TC tablets.
Trade marks are owned by or licensed to the ViiV Healthcare group of companies.
3TC 150 mg tablets: AUST R 54313
3TC 300 mg tablets: AUST R 81359
© 2021 ViiV Healthcare group of companies or its licensor.
Version 7.0
Published by MIMS December 2021
 A reduction in the dose and/or an increase in the dosing interval should be considered in children aged at least three months and weighing less than 25 kg.
A reduction in the dose and/or an increase in the dosing interval should be considered in children aged at least three months and weighing less than 25 kg. Intermittent dialysis is unlikely to require further dose modification from that defined by creatinine clearance.
Intermittent dialysis is unlikely to require further dose modification from that defined by creatinine clearance. Common laboratory abnormalities observed during therapy are listed in Table 4.
Common laboratory abnormalities observed during therapy are listed in Table 4. Lamivudine appears to be well tolerated and most serious adverse events reported in clinical trials are not considered to be drug related. Adverse reactions from the 4 pivotal studies in adult patients receiving the recommended dose of lamivudine (150 mg bd) in combination with zidovudine 600 mg/day are included in Table 5 together with serious adverse reactions reported in large scale open studies.
Lamivudine appears to be well tolerated and most serious adverse events reported in clinical trials are not considered to be drug related. Adverse reactions from the 4 pivotal studies in adult patients receiving the recommended dose of lamivudine (150 mg bd) in combination with zidovudine 600 mg/day are included in Table 5 together with serious adverse reactions reported in large scale open studies. Cases of pancreatitis have occurred rarely in adult patients and more commonly in children. However, it is not clear whether these cases were due to drug treatment or to their underlying HIV disease. Treatment with lamivudine should be stopped immediately if clinical signs or symptoms or laboratory abnormalities suggestive of pancreatitis occur.
Cases of pancreatitis have occurred rarely in adult patients and more commonly in children. However, it is not clear whether these cases were due to drug treatment or to their underlying HIV disease. Treatment with lamivudine should be stopped immediately if clinical signs or symptoms or laboratory abnormalities suggestive of pancreatitis occur. See Table 7.
See Table 7. The data from the clinical trials have shown no significant difference in the frequency or severity of unwanted effects between administration of 150 mg twice daily and 300 mg once daily. There was, however, a higher incidence of hyperamylasaemia in the once daily regimen.
The data from the clinical trials have shown no significant difference in the frequency or severity of unwanted effects between administration of 150 mg twice daily and 300 mg once daily. There was, however, a higher incidence of hyperamylasaemia in the once daily regimen. Selected laboratory abnormalities experienced by therapy naive (≤ 56 days of antiretroviral therapy) paediatric patients are listed in Table 9.
Selected laboratory abnormalities experienced by therapy naive (≤ 56 days of antiretroviral therapy) paediatric patients are listed in Table 9.

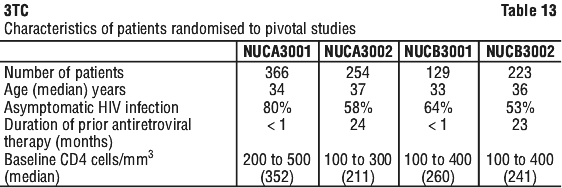
 The data showed there was a significant reduction in progression to the combined endpoint of a new AIDS event or death for patients who received lamivudine in combination with zidovudine containing regimens compared to patients maintained on zidovudine containing regimens alone (p < 0.0001). The hazard ratio (HR) was 0.427 (95% confidence interval, 0.318 - 0.572) or a 57% reduction in risk. In addition, the data indicated a significant reduction in death, regardless of causality, in the combination lamivudine plus zidovudine containing regimens as compared to the zidovudine containing regimens alone (p = 0.0007); HR = 0.399 (95% CI, 0.230 - 0.693) or a 60% reduction in risk.
The data showed there was a significant reduction in progression to the combined endpoint of a new AIDS event or death for patients who received lamivudine in combination with zidovudine containing regimens compared to patients maintained on zidovudine containing regimens alone (p < 0.0001). The hazard ratio (HR) was 0.427 (95% confidence interval, 0.318 - 0.572) or a 57% reduction in risk. In addition, the data indicated a significant reduction in death, regardless of causality, in the combination lamivudine plus zidovudine containing regimens as compared to the zidovudine containing regimens alone (p = 0.0007); HR = 0.399 (95% CI, 0.230 - 0.693) or a 60% reduction in risk.


 The data demonstrate that the proportion of patients with plasma HIV-1 RNA < 400 copies/mL at week 24 did not differ between treatment groups (od: 72%; bid: 72%). While the subjects in the od group had a baseline plasma HIV-1 RNA level 0.8 log10 copies/mL higher than those of the bid group (od: 5.1 log10 copies/mL; bid: 4.3 log10 copies/mL), the two groups were comparable with respect to on treatment plasma HIV-1 RNA response. Twenty-nine (5%) of subjects had virological failure by week 24 but virus for resistance testing could only be isolated from 22 samples (11 subjects in each arm); the incidence of resistance was comparable in the treatment arms.
The data demonstrate that the proportion of patients with plasma HIV-1 RNA < 400 copies/mL at week 24 did not differ between treatment groups (od: 72%; bid: 72%). While the subjects in the od group had a baseline plasma HIV-1 RNA level 0.8 log10 copies/mL higher than those of the bid group (od: 5.1 log10 copies/mL; bid: 4.3 log10 copies/mL), the two groups were comparable with respect to on treatment plasma HIV-1 RNA response. Twenty-nine (5%) of subjects had virological failure by week 24 but virus for resistance testing could only be isolated from 22 samples (11 subjects in each arm); the incidence of resistance was comparable in the treatment arms. The data demonstrate that there were similar increases in median CD4+ cell counts observed between 3TC treatment groups at week 24 (OD: 128 cells/mm3; BID: 123 cells/mm3).
The data demonstrate that there were similar increases in median CD4+ cell counts observed between 3TC treatment groups at week 24 (OD: 128 cells/mm3; BID: 123 cells/mm3). Although there was a small numerical difference in treatment response in favour of the control group, this difference was primarily driven by a slightly higher incidence of treatment discontinuations in the OAD arms for reasons other than virological failure. Fifteen (10%) subjects had virological failure associated with resistance mutations; the incidence was comparable in the treatment arms.
Although there was a small numerical difference in treatment response in favour of the control group, this difference was primarily driven by a slightly higher incidence of treatment discontinuations in the OAD arms for reasons other than virological failure. Fifteen (10%) subjects had virological failure associated with resistance mutations; the incidence was comparable in the treatment arms. The peak increase in CD4 count was 104 cells/mm3 at week 4 in Arm A and 6 cells/mm3 in Arm B. CD4 counts were maintained at or close to baseline values in both arms of the study to week 48. Serum HIV-1 RNA concentrations decreased during the same period and for the subset of patients with > 1000 copies/mL at baseline was significantly different to baseline in Arm A at week 48 (-0.7 log10, p = 0.031), and in Arm B at week 12 (-0.2 log10, p = 0.009), week 24 (-0.2 log10, p = 0.008) and week 48 (0.2 log10, p = 0.032).
The peak increase in CD4 count was 104 cells/mm3 at week 4 in Arm A and 6 cells/mm3 in Arm B. CD4 counts were maintained at or close to baseline values in both arms of the study to week 48. Serum HIV-1 RNA concentrations decreased during the same period and for the subset of patients with > 1000 copies/mL at baseline was significantly different to baseline in Arm A at week 48 (-0.7 log10, p = 0.031), and in Arm B at week 12 (-0.2 log10, p = 0.009), week 24 (-0.2 log10, p = 0.008) and week 48 (0.2 log10, p = 0.032).
 This study demonstrated a significant benefit of combination zidovudine/lamivudine therapy over didanosine monotherapy by clinical and laboratory measures.
This study demonstrated a significant benefit of combination zidovudine/lamivudine therapy over didanosine monotherapy by clinical and laboratory measures. The abacavir/lamivudine once daily dosing group was demonstrated to be non-inferior to the twice daily group according to the prespecified non-inferiority margin of -12%, for the primary endpoint of < 80 copies/mL at Week 48 as well as at Week 96 (secondary endpoint) and all other thresholds tested (< 200 copies/mL, < 400 copies/mL, < 1000 copies/mL), which all fell well within this non-inferiority margin. Subgroup analyses testing for heterogeneity of once vs twice daily demonstrated no significant effect of sex, age, or viral load at randomisation. Conclusions supported non-inferiority regardless of analysis method.
The abacavir/lamivudine once daily dosing group was demonstrated to be non-inferior to the twice daily group according to the prespecified non-inferiority margin of -12%, for the primary endpoint of < 80 copies/mL at Week 48 as well as at Week 96 (secondary endpoint) and all other thresholds tested (< 200 copies/mL, < 400 copies/mL, < 1000 copies/mL), which all fell well within this non-inferiority margin. Subgroup analyses testing for heterogeneity of once vs twice daily demonstrated no significant effect of sex, age, or viral load at randomisation. Conclusions supported non-inferiority regardless of analysis method. Genotypic resistance analyses were conducted on samples with plasma HIV-1 RNA > 1000 copies/mL. More cases of resistance were detected among patients who had received lamivudine solution, in combination with other antiretroviral solutions, compared with those who received similar doses of tablet formulation. This is consistent with the lower rates of antiviral suppression observed in these patients (see Section 5.2 Pharmacokinetic Properties, Pharmacokinetics in children).
Genotypic resistance analyses were conducted on samples with plasma HIV-1 RNA > 1000 copies/mL. More cases of resistance were detected among patients who had received lamivudine solution, in combination with other antiretroviral solutions, compared with those who received similar doses of tablet formulation. This is consistent with the lower rates of antiviral suppression observed in these patients (see Section 5.2 Pharmacokinetic Properties, Pharmacokinetics in children). Plasma lamivudine concentrations following oral solution administration were obtained in a subset of children who were enrolled in the full ARROW study. The ARROW PK Substudy 2 was designed to compare the plasma pharmacokinetics of lamivudine when co-administered as oral solution versus scored tablet (n = 19) of HIV-1 infected children (weighing 12 to 15 kg and aged 2 to 4 years) who were enrolled in the full ARROW study. The children were receiving lamivudine oral solution (twice daily) and were ready to switch to tablet form (twice daily). For lamivudine, the oral solution dose was 60 mg twice daily and the tablet dose was 75 mg twice daily (½ of a scored lamivudine-zidovudine tablet). Serial pharmacokinetic samples were obtained after at least 24 weeks on solution formulation and again at 4 weeks after switching to the tablet formulation. The plasma exposures of lamivudine were higher following the administration of lamivudine as the lamivudine-zidovudine tablet with dose normalised Cmax and AUC(0-t) values approximately 55% and 58% higher, respectively, compared to the lamivudine oral solution.
Plasma lamivudine concentrations following oral solution administration were obtained in a subset of children who were enrolled in the full ARROW study. The ARROW PK Substudy 2 was designed to compare the plasma pharmacokinetics of lamivudine when co-administered as oral solution versus scored tablet (n = 19) of HIV-1 infected children (weighing 12 to 15 kg and aged 2 to 4 years) who were enrolled in the full ARROW study. The children were receiving lamivudine oral solution (twice daily) and were ready to switch to tablet form (twice daily). For lamivudine, the oral solution dose was 60 mg twice daily and the tablet dose was 75 mg twice daily (½ of a scored lamivudine-zidovudine tablet). Serial pharmacokinetic samples were obtained after at least 24 weeks on solution formulation and again at 4 weeks after switching to the tablet formulation. The plasma exposures of lamivudine were higher following the administration of lamivudine as the lamivudine-zidovudine tablet with dose normalised Cmax and AUC(0-t) values approximately 55% and 58% higher, respectively, compared to the lamivudine oral solution.
