What is in this leaflet
This leaflet answers some common questions about ACTIKERALL.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using ACTIKERALL against the benefits they expect it will have for you.
If you have any concerns about using this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What ACTIKERALL is used for
ACTIKERALL is used to treat mild to moderate solar keratosis lesions, also known as actinic keratosis lesions or sunspots.
Solar keratoses are rough, red, scaly or crusty spots on the skin that are caused by too much exposure to sunlight. They are more common on sun-exposed areas, such as the face, nose, ears, chest, forearms and back of the hands.
Solar keratoses are usually harmless, but there is a risk that they may turn into skin cancer.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is available only with a doctor's prescription.
How it works
ACTIKERALL contains two active substances, fluorouracil and salicylic acid.
Fluorouracil belongs to a group of medicines known as antimetabolites which inhibit the growth of cells (cytostatic agent).
Salicylic acid is a hard skin softening substance.
Before you use ACTIKERALL
When you must not use it
Do not use ACTIKERALL if you have an allergy to:
- any medicine containing fluorouracil, salicylic acid or acetylsalicylic acid (aspirin)
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
Do not use this medicine if you have dihydropyrimidine dehydrogenase (DPD) enzyme deficiency or are taking medicines that could lower your DPD levels. This enzyme plays an important role in the breakdown of fluorouracil. As a result, fluorouracil might accumulate in your body. It may be necessary to have your DPD levels checked before starting treatment with ACTIKERALL.
Do not use ACTIKERALL if you have kidney problems.
Do not use this medicine if you are pregnant or there is a possibility you may be pregnant. It may affect your developing baby if you use it during pregnancy.
Do not breast-feed if you are using this medicine. It is unknown whether the active ingredient fluorouracil passes into breast milk. Tell your doctor if you are pregnant or plan to become pregnant or are breast-feeding.
Your doctor can discuss with you the risks and benefits involved.
Do not use this medicine after the expiry date (EXP) printed on the pack, if the packaging is torn, shows signs of tampering, or 3 months has passed after first opening the bottle. If it has expired or is damaged, return it to your pharmacist for disposal.
Once opened, the solution can only be used for 3 months. Discard 3 months after first opening the bottle.
If you are not sure whether you should start using ACTIKERALL, talk to your doctor or pharmacist first.
Before you use it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
ACTIKERALL contains the ingredient dimethyl sulfoxide which may be an irritant to the skin.
Tell your doctor if you have or have had any inflammatory skin conditions, bleeding skin lesions or any other skin condition.
You must tell your doctor if you are using any other dermatological products to treat solar keratoses.
Tell your doctor if you have or have had DPD (dihydropyrimidine dehydrogenase) enzyme deficiency.
Tell your doctor if you suffer from a reduced ability to sense touch, pain and temperature. If you have diabetes you may experience these symptoms. In this case, your treatment areas will be closely monitored by your doctor.
Tell your doctor if you work outside for long periods of time during the day. ACTIKERALL is not recommended for people who work outdoors for long periods of time.
If you have not told your doctor about any of the above, tell them before you start using ACTIKERALL.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and ACTIKERALL may interfere with each other. These include:
- medicines used to treat viruses such as chicken pox or shingles
- phenytoin, a medicine used to treat epilepsy
- methotrexate, a medicine used to treat cancer and autoimmune diseases
- medicines used to treat diabetes e.g. sulphonylureas.
These medicines may be affected by ACTIKERALL or may affect how well it works. You may need different amounts of your medicine, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while using this medicine.
How to use ACTIKERALL
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the carton/bottle, ask your doctor or pharmacist for help.
When to use it
ACTIKERALL should be applied once daily unless your doctor has told you otherwise.
If you have a solar keratosis in an area of thin skin e.g. around the eyes and temple, your doctor may tell you to apply ACTIKERALL less frequently. If severe side effects occur, your doctor may reduce the frequency of drug application to three times per week until the side effects improve.
It may also be necessary for your doctor to monitor your treatment more often.
How long to use it for
ACTIKERALL is applied to the treatment area once daily until the lesions have completely cleared or for up to a maximum of 12 weeks.
Improvement of solar keratosis can be seen as early as 4 weeks after starting treatment and the improvement increases over time up to 12 weeks.
- The clearance of solar keratoses may continue for up to 8 weeks after you have stopped using ACTIKERALL.
- If you feel that the product is not working as described, talk to your doctor or pharmacist.
How to use it
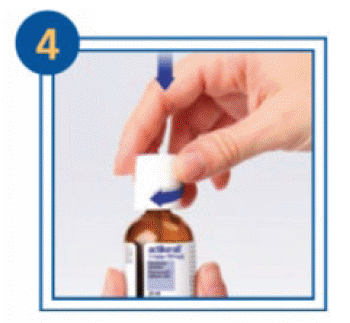
For the first application to the skin (topical use):
- To open the bottle, press the lid down and turn.

- Remove excess solution from the brush by wiping it on the neck of the bottle.

- Dab the solution on the solar keratosis and a small area of healthy skin surrounding it once daily. This rim of healthy skin should be no more than 0.5 cm wide. Do not apply on a total area greater than 25 square centimetres (cm2) at any one time.
Multiple solar keratoses (up to 10 lesions) can be treated simultaneously, but do not use ACTIKERALL on large areas of skin. The total area of skin being treated with ACTIKERALL at any one time should not exceed 25 cm2. As a guide, the rectangle outline drawn on the side of the medicine carton, where your pharmacist may attach a dispensing label, represents a total area of 25 cm2. Examples of acceptable treatment areas are below:
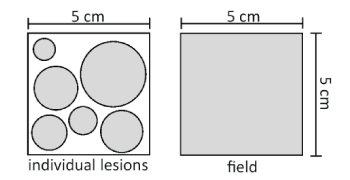
When treating individual solar keratosis lesions, ensure the solution only comes into contact with the lesion and a maximum rim of 0.5 cm of healthy skin around the lesion.
If your doctor has advised you to use ACTIKERALL as field treatment, multiple lesions as well as the surrounding skin can be treated at the same time within the treatment area of up to 25 cm2.
Let the solution dry to form a film.
Do not cover with a dressing.
- Close the bottle properly.
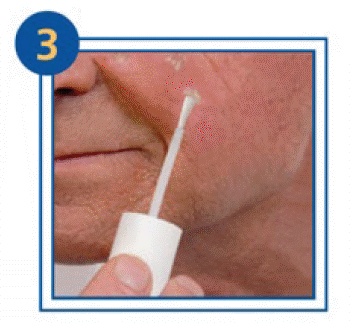
Subsequent applications:
- Remove the white film on your skin from the previous day’s application by simply peeling it off. Warm water may help to remove the film.
- Follow the instructions as described for the first application.
Further instructions
ACTIKERALL must not be allowed to come into contact with the eyes, the inside of the mouth or nose or the genitals (mucous membranes).
ACTIKERALL should not be applied to hairy skin as the hair becomes stuck together.
Hair removal such as shaving should be considered prior to applying ACTIKERALL.
ACTIKERALL may permanently stain clothing, fabric or acrylics (such as acrylic bathtubs), so avoid contact with them.
ACTIKERALL is flammable: keep away from fire or flames.
Close the bottle tightly to prevent it from drying out. If ACTIKERALL dries out, it must not be used any longer.
Do not use ACTIKERALL if you notice any crystals.
Do not use ACTIKERALL 3 months after first opening the bottle.
If you forget to use it
Do not apply more than once daily. You will be more likely to experience skin reactions and they will be more severe if used more than recommended.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to use your medicine, ask your pharmacist for some suggestions.
If you use too much (overdose) or swallow the solution
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else has used too much ACTIKERALL or in the case of accidental swallowing. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are using ACTIKERALL
Your appearance may look worse and you may feel uncomfortable symptoms on the treated area while treatment is in progress. Sometimes it can take several weeks after treatment with ACTIKERALL has stopped before you see any improvement in your condition.
Things you must do
Limit your exposure to the sun during treatment with ACTIKERALL. This will help reduce any side effects of the medication.
If you are about to start any new medicine, remind your doctor and pharmacist that you are using ACTIKERALL.
Tell any other doctors, dentists, and pharmacists who treat you that you are using this medicine.
If you become pregnant while using this medicine, tell your doctor immediately.
Things you must not do
Do not use ACTIKERALL to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop using your medicine without checking with your doctor.
Do not let ACTIKERALL come into contact with the eyes, the inside of the mouth or nose or the genitals (mucous membranes).
Things to be careful of
While treatment is in progress, it is important to consider the following:
- Wherever possible, the skin area treated with ACTIKERALL should be protected against direct sunlight and other forms of ultraviolet radiation using hats and clothing. The treated skin should not be covered with dressings or bandages.
- Before going outdoors, apply a broad-spectrum sunscreen (SPF 30 or higher) over the dried film of ACTIKERALL on the lesion if the skin surface is unbroken (not eroded, scabby, bleeding or exfoliated). Reapply the sunscreen several times during the day.
- ACTIKERALL should not be used on bleeding lesions.
- ACTIKERALL should not be used around the eyes, nose or mouth unless directed by your doctor.
- Avoid cosmetics and other dermatological preparations on the treatment area, unless otherwise instructed by your doctor. Once you stop using ACTKERALL, talk to your doctor or pharmacist before applying cosmetics or other dermatological products on the affected skin.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are using ACTIKERALL.
This medicine helps most people with solar keratoses, but it can have unwanted side effects. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
The most common side effects of ACTIKERALL are reactions at the application site and include:
- redness
- inflammation
- irritation
- itching
- burning
- pain
- crusting
- whitish discolouration and scaling of the skin
- rash
- scarring
- bleeding in the treatment area
Tell your doctor if you experience any of the above side effects and they worry you. These reactions are common and are signs that the medicine is working.
The salicylic acid in ACTIKERALL may cause slight irritation, such as skin inflammation (dermatitis) and contact allergic reactions. Symptoms of contact allergic reactions may include itching, reddening and small blisters even outside the area of application.
Tell your doctor if you experience an allergic reaction that sounds like this.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- swelling and soreness of the mouth and tongue.
- fever and chills
These are serious side effects. You may need urgent medical attention or hospitalisation. These side effects are rare.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above such as headache may also occur in some people.
After using ACTIKERALL
Storage
Keep this medicine in a cool, dry place where the temperature stays below 25°C. Do not refrigerate. Do not freeze. Keep the bottle tightly closed.
Do not store ACTIKERALL or any other medicine in the bathroom or near a sink. Do not leave it on a windowsill or in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a half metres above the ground is a good place to store medicines.
Do not use ACTIKERALL after the expiry date which is stated on the label and on the carton.
Disposal
If your doctor tells you to stop using this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
ACTIKERALL is a clear, colourless to slightly orange–white solution. It is packed in a 25 mL brown glass bottle with a child-resistant closure.
Ingredients
The active substances of ACTIKERALL are fluorouracil and salicylic acid.
One gram of solution contains:
- 5 mg of fluorouracil, and
- 100 mg of salicylic acid.
It also contains the following inactive excipients:
- dimethyl sulfoxide
- ethanol absolute
- ethyl acetate
- pyroxylin
- butyl methacrylate/ methyl methacrylate copolymer (3:1)
This medicine does not contain lactose, sucrose, gluten, tartazine or any other azo dyes.
Sponsor
ACTIKERALL is distributed by:
Mayne Pharma International Pty Ltd
1538 Main North Road
Salisbury South, SA 5106
Under licence from Almirall
® = Registered Trademark
Australian Registration Number:
AUST R 287446
This leaflet was prepared in March 2023.
For the most up to date version of this leaflet go to actikerall.com.au
Published by MIMS June 2023
 The adverse events reported in the Phase III field directed study (no. 98605101-1401) are consistent with those described above. In the Phase II comparator controlled study (no. H 1005 6002-1007), the most common local skin reactions were erythema, scab/crusting and burning, mainly of mild to moderate intensity.
The adverse events reported in the Phase III field directed study (no. 98605101-1401) are consistent with those described above. In the Phase II comparator controlled study (no. H 1005 6002-1007), the most common local skin reactions were erythema, scab/crusting and burning, mainly of mild to moderate intensity.
 Patients had 4 -10 clinically confirmed solar keratosis lesions on the face, forehead, and/or bald scalp. The total treatment area was up to a maximum of 25 cm2. Patients were treated for a maximum of 12 weeks or until the lesions had completely cleared or ulceration of the lesions had occurred.
Patients had 4 -10 clinically confirmed solar keratosis lesions on the face, forehead, and/or bald scalp. The total treatment area was up to a maximum of 25 cm2. Patients were treated for a maximum of 12 weeks or until the lesions had completely cleared or ulceration of the lesions had occurred. Data for overall lesion response at weeks 2, 6, the end of the trial (week 12) and at the posttreatment assessment shown on Figure 3.
Data for overall lesion response at weeks 2, 6, the end of the trial (week 12) and at the posttreatment assessment shown on Figure 3. By EOT, the investigators rated the global outcome (PGA) as very good or good in 82.3% of the patients in the Actikerall group (144 patients), in 74.3% of patients in the Solaraze group (130 patients) and 65.6% of patients in the placebo group (61 patients).
By EOT, the investigators rated the global outcome (PGA) as very good or good in 82.3% of the patients in the Actikerall group (144 patients), in 74.3% of patients in the Solaraze group (130 patients) and 65.6% of patients in the placebo group (61 patients). From study initiation to 6-month-post-treatment follow-up, recurrence of cleared solar keratosis lesions (no clinically visible lesions in the treatment area on Day 98) was reported in 39.4% (13/33) of patients receiving Actikerall and 84.8% (28/33) of patients receiving cryotherapy (see Figure 4).
From study initiation to 6-month-post-treatment follow-up, recurrence of cleared solar keratosis lesions (no clinically visible lesions in the treatment area on Day 98) was reported in 39.4% (13/33) of patients receiving Actikerall and 84.8% (28/33) of patients receiving cryotherapy (see Figure 4). In patients with complete clinical clearance at 14 weeks post-treatment, 27.3% (3/11) of patients receiving Actikerall and 50.0% (4/8) of patients receiving cryotherapy reported recurrent lesions at 6-month post-treatment follow-up.
In patients with complete clinical clearance at 14 weeks post-treatment, 27.3% (3/11) of patients receiving Actikerall and 50.0% (4/8) of patients receiving cryotherapy reported recurrent lesions at 6-month post-treatment follow-up. Molecular formula: C4H3FN2O2.
Molecular formula: C4H3FN2O2. Molecular formula: C7H6O3.
Molecular formula: C7H6O3.