1 Name of Medicine
Afqlir (aflibercept) 2 mg is a biosimilar medicine to Eylea (aflibercept) 2 mg. The comparability of Afqlir (aflibercept) with Eylea (aflibercept) has been demonstrated with regard to physicochemical characteristics and efficacy and safety outcomes (see Section 5.1 Pharmacodynamic Properties, Clinical trials; Section 4.8 Adverse Effects (Undesirable Effects)). The evidence for comparability supports the use of Afqlir for the listed indications.
2 Qualitative and Quantitative Composition
Aflibercept is a recombinant fusion protein consisting of portions of human VEGF receptor 1 and 2 extracellular domains fused to the Fc portion of human IgG1. Aflibercept is produced in Chinese hamster ovary (CHO) K1 cells by recombinant DNA technology.
For the full list of excipients, see Section 6.1 List of Excipients.
Afqlir 40 mg/mL (vial for 2 mg dosing).
Each 1 mL of Afqlir solution contains 40 mg aflibercept. Each vial contains approximately 240 microL of solution. This amount is sufficient to deliver a single dose of 50 microL solution for intravitreal injection containing 2 mg aflibercept.
Afqlir 40 mg/mL (pre-filled syringe for 2 mg dosing).
Each 1 mL of Afqlir solution contains 40 mg aflibercept. Each pre-filled syringe contains approximately 165 microL of solution. This amount is sufficient to deliver a single dose of 50 microL solution for intravitreal injection containing 2 mg aflibercept.
2 mg is the only dose of Afqlir available. There is no 8 mg Afqlir available.3 Pharmaceutical Form
Solution for intravitreal injection.
Afqlir 40 mg/mL is a sterile, clear, colourless to slightly brownish-yellow, preservative-free, iso-osmotic aqueous solution.
4.1 Therapeutic Indications
Afqlir (aflibercept) 2 mg is indicated in adults for the treatment of:
neovascular (wet) age-related macular degeneration (wet AMD);
visual impairment due to macular oedema secondary to central retinal vein occlusion (CRVO);
visual impairment due to macular oedema secondary to branch retinal vein occlusion (BRVO);
diabetic macular oedema (DME);
visual impairment due to myopic choroidal neovascularisation (myopic CNV).
4.2 Dose and Method of Administration
Afqlir is for intravitreal injection only.
It must only be administered by a qualified ophthalmologist experienced in administering intravitreal injections.
Dosage.
The recommended dose for Afqlir (40 mg/mL) is 2 mg aflibercept, equivalent to an injection volume of 50 microL.
Do not administer the 8 mg dosing of aflibercept using Afqlir. There is no 8 mg Afqlir available. If the 8 mg dosing is required, other aflibercept products offering such an option should be used.
The interval between doses injected into the same eye should not be shorter than one month.
Advice on treatment initiation and maintenance of therapy specific to each patient population is described in the section below. Once optimal visual acuity is achieved and/or there are no signs of disease activity, treatment may then be continued with a treat-and-extend regimen with gradually increased treatment intervals to maintain stable visual and/or anatomic outcomes. If disease activity persists or recurs, the treatment interval may be shortened accordingly. Monitoring should be done at injection visits. The monitoring and treatment schedule should be determined by the treating ophthalmologist based on the individual patient's response. If visual and anatomic outcomes indicate that the patient is not benefiting from continued treatment, Afqlir should be discontinued.
Treatment of neovascular (wet) age-related macular degeneration (wet AMD).
Afqlir 2 mg treatment is initiated with one Afqlir 2 mg injection per month for three consecutive months, followed by one injection every two months.
Based on the ophthalmologist's judgement of visual and/or anatomic outcomes, the treatment interval may be maintained at two months or further extended using a treat-and-extend dosing regimen, by increasing injection intervals in 2- or 4-weekly increments while maintaining stable visual and/or anatomic outcomes. If visual and/or anatomic outcomes deteriorate, the treatment interval should be shortened to a minimum of four weeks based on anatomical and/or visual outcomes.
Generally, once optimal visual acuity is achieved and/or there are no signs of disease activity, the treatment interval may be adjusted based on visual and/or anatomic outcomes.
Treatment intervals greater than four months (16 weeks) between injections have not been studied (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Treatment of visual impairment due to macular oedema secondary to central retinal vein occlusion (CRVO).
Afqlir 2 mg treatment is initiated with one Afqlir 2 mg injection per month for three consecutive months. After the first three monthly injections, the treatment interval may be adjusted based on visual and/or anatomic outcomes.
Treatment of visual impairment due to macular oedema secondary to branch retinal vein occlusion (BRVO).
Afqlir 2 mg treatment is initiated with one Afqlir 2 mg injection per month for three consecutive months. After the first three monthly injections, the treatment interval may be adjusted based on visual and/or anatomic outcomes.
Treatment of diabetic macular oedema (DME).
Afqlir 2 mg treatment is initiated with one Afqlir 2 mg injection per month for five consecutive months.
Following the initiation period and based on the ophthalmologist's judgement of visual and/or anatomic outcomes, the treatment interval may then be maintained at an injection every two months or further individualised, such as with a treat-and-extend dosing regimen, by increasing injection intervals in 2- or 4-weekly increments while maintaining stable visual and/or anatomic outcomes. If visual and/or anatomic outcomes deteriorate, the treatment interval should be shortened accordingly. Treatment intervals shorter than 4 weeks or longer than 4 months have not been studied (see Section 5.1 Pharmacodynamic Properties, Clinical trials).
Treatment of visual impairment due to myopic choroidal neovascularisation (myopic CNV).
Afqlir 2 mg treatment is initiated with one Afqlir 2 mg injection (equivalent to 50 microL).
Additional doses should be administered only if visual and/or anatomic outcomes indicate that the disease persists. Recurrences are treated like a new manifestation of the disease.
Method of administration.
Intravitreal injections must be carried out according to medical standards and applicable guidelines by a qualified ophthalmologist experienced in administering intravitreal injections. In general, adequate anaesthesia and asepsis, including topical broad-spectrum microbicide, have to be ensured. Surgical hand disinfection, sterile gloves, a sterile drape, and a sterile eyelid speculum (or equivalent) are recommended.
Immediately following the intravitreal injection, patients should be monitored for elevation in intraocular pressure. Appropriate monitoring may consist of a check for perfusion of the optic nerve head or tonometry. If required, sterile equipment for paracentesis should be available.
Following intravitreal injection patients should be instructed to report any symptoms suggestive of endophthalmitis (e.g. eye pain, redness of the eye, photophobia, blurring of vision) without delay.
Each pre-filled syringe or vial should only be used for the treatment of a single eye.
The recommended dose is 2 mg aflibercept (equivalent to 50 microL solution for injection). The pre-filled syringe and the glass vial contain more than this recommended dose. Therefore, the excess volume must be expelled before injecting (see Instructions for use/ handling). Injecting the entire volume of the glass vial or the pre-filled syringe could result in overdose.
Afqlir 40 mg/mL (vial for 2 mg dosing).
To administer 2 mg aflibercept (equivalent to 50 microL solution for injection), eliminate all bubbles and expel excess drug by slowly depressing the plunger so that the flat plunger edge aligns with the line that marks 0.05 mL (equivalent to 50 microL) on the syringe before injecting.
Afqlir 40 mg/mL (pre-filled syringe for 2 mg dosing).
To administer 2 mg aflibercept (equivalent to 50 microL solution for injection), eliminate all bubbles and expel excess drug by slowly depressing the plunger to align the plunger dome edge (not the tip of the dome) with the black dosing line on the syringe. This will ensure a delivery equivalent to 50 microL i.e. 2 mg aflibercept.
After injection any unused product or waste material must be discarded.
Instructions for use/ handling.
Vial. The vial contains more than the recommended dose of 2 mg aflibercept (equivalent to 0.05 mL). The excess volume must be discarded prior to administration.
Storage and inspection.
Store Afqlir in the refrigerator at 2°C - 8°C; do not freeze. Keep the vial in the outer carton to protect from light.
Prior to use, the unopened vial of Afqlir may be kept at room temperature below 30°C for up to 14 days. Store in original carton and do not open vial until time of use. After opening the vial, proceed under aseptic conditions.
Afqlir is a clear and colourless to slightly brownish-yellow solution.
Afqlir should be inspected visually for any particulates, cloudiness and/or discoloration or any variation in physical appearance prior to administration. In the event of any of these being observed, discard Afqlir.
Do not use if the packaging, vial and/or filter needle are damaged or expired.
Preparation and administration.
Each glass vial is for one-time use in one eye only.
For preparation and intravitreal injection the following single use medical devices are needed:
A 5-micrometer blunt filter needle (18 G x 1½ inch), sterile, supplied with the vial.
A 1 mL Luer-lock syringe with a 0.05 mL dose mark, sterile (not supplied).
For the intravitreal injection, a sterile 30 G x ½ inch injection needle should be used (not supplied).
Use aseptic technique to carry out the following steps.
Injection procedure.
1. Remove the protective plastic cap from the vial.
2. Clean the top of the vial with an alcohol wipe.
3. Attach the supplied 18 G x 1½ inch, 5-micron filter needle to a 1 mL sterile, Luer-lock syringe by twisting it onto the syringe tip.
4. Push the filter needle into the centre of the vial stopper until the needle is completely inserted into the vial and the tip touches the bottom or bottom edge of the vial.
5. Using aseptic technique withdraw all of the Afqlir vial content into the syringe, keeping the vial in an upright position, slightly inclined to ease complete withdrawal. To deter the introduction of air, ensure the bevel of the filter needle is submerged into the liquid. Continue to tilt the vial during withdrawal keeping the bevel of the filter needle submerged in the liquid.
6. Ensure that the plunger rod is drawn sufficiently back when emptying the vial in order to completely empty the filter needle.
7. Remove the filter needle from the syringe and properly dispose of the filter needle.
Note.
Filter needle is not to be used for intravitreal injection.
8. Attach the 30 G x ½ inch injection needle to the syringe by firmly twisting the injection needle onto the Luer-lock syringe tip.
Carefully remove the needle cap by pulling it straight off.
9. Holding the syringe with the needle pointing up, check the syringe for bubbles. If there are bubbles, gently tap the syringe with your finger until the bubbles rise to the top.
10. To eliminate all of the bubbles and to expel excess drug, slowly depress the plunger so that the flat plunger edge aligns with the line that marks 0.05 mL on the syringe.
Note.
Inject immediately after preparation.
11. Inject slowly until the rubber stopper reaches the end of the syringe to deliver the volume of 0.05 mL. Confirm delivery of the full dose by checking that the rubber stopper has reached the end of the syringe barrel.
12. The vial is for one-time use in one eye only. Do not extract multiple doses from a single vial, as this may increase the risk of contamination and subsequent infection. Any unused medicinal product or waste material should be disposed of in accordance with local requirements.
Pre-filled syringe. The pre-filled syringe contains more than the recommended dose of 2 mg aflibercept (equivalent to 0.05 mL). The excess volume must be discarded prior to administration.
Storage and inspection.
Store Afqlir in the refrigerator at 2°C - 8°C; do not freeze. Keep the pre-filled syringe in the outer carton to protect from light.
Prior to usage, the unopened blister of Afqlir may be stored at room temperature below 30°C for up to 14 days. Store in original carton and do not open sealed blister pack until time of use. After opening the blister, proceed under aseptic conditions.
Afqlir is a clear and colourless to slightly brownish-yellow solution.
The solution should be inspected visually for any particulates, cloudiness and/or discoloration or any variation in physical appearance prior to administration. In the event of any of these being observed, discard the medicinal product.
Do not use if the package is open or damaged. Do not use if any part of the pre-filled syringe is damaged, if the syringe cap is detached from the Luer lock, or if the pre-filled syringe is expired.
Preparation and administration.
Each pre-filled syringe is for one-time use in one eye only.
Do not open the sterile pre-filled syringe blister outside the clean administration room.
For the intravitreal injection, a sterile 30 G x ½ inch injection needle should be used (not supplied).
Use aseptic technique to carry out the following steps.
Injection procedure.
1. When ready to administer Afqlir, open the carton and remove sterilised blister pack. Carefully peel open the sterilised blister pack ensuring the sterility of its contents. Keep the syringe in the sterile tray until you are ready for assembly.
2. Using aseptic technique, remove the syringe from the sterilised blister pack.
3. To remove the syringe cap, hold the syringe in one hand while using the other hand to grasp the syringe cap with the thumb and forefinger. Snap off (do not turn or twist) the syringe cap.
Note: To avoid compromising the sterility of the product, do not pull back on the plunger.
4. Using aseptic technique, firmly twist a 30 G x ½ inch injection needle onto the Luer-lock syringe tip.
Carefully remove the needle cap by pulling it straight off.
5. Holding the syringe with the needle pointing up, check the syringe for bubbles. If there are bubbles, gently tap the syringe with your finger until the bubbles rise to the top.
6. Eliminate all bubbles and expel excess drug by slowly depressing the plunger rod to align the plunger dome edge (not the tip of the dome) with the black dosing line on the syringe (equivalent to 50 microlitres).
Note.
Inject immediately after priming the syringe.
7. Inject slowly until the rubber stopper reaches the end of the syringe to deliver the volume of 0.05 mL. Confirm delivery of the full dose by checking that the rubber stopper has reached the end of the syringe barrel. Do not apply additional pressure once the end of the syringe is reached.
8. The pre-filled syringe is for one-time use in one eye only. Do not extract multiple doses from a pre-filled syringe, as this may increase the risk of contamination and subsequent infection. Any unused product or waste material should be disposed of in accordance with local requirements.
Dosage adjustment.
Patients with hepatic and/or renal impairment.
No specific studies in patients with hepatic and/or renal impairment were conducted with aflibercept. Available data do not suggest a need for a dose adjustment with aflibercept in these patients (see Section 5.2 Pharmacokinetic Properties).
For aflibercept 2 mg, pharmacokinetic analysis of patients with wet AMD in the VIEW 2 study, of which 40% had renal impairment (24% mild, 15% moderate, and 1% severe), revealed no differences with respect to plasma concentrations of active drug after intravitreal administration every 4 or 8 weeks. Similar results were seen in patients with CRVO in the GALILEO study, with DME in the VIVIDDME study and with myopic CNV in the MYRROR study.
Use in elderly.
Available data do not suggest a need for a dose adjustment with aflibercept 2 mg in these patients. (See Section 5.1 Pharmacodynamic Properties, Clinical trials.)4.3 Contraindications
Known hypersensitivity to aflibercept or to any of the excipients of Afqlir (see Section 6.1 List of Excipients).
Ocular or periocular infection.
Active severe intraocular inflammation.
4.4 Special Warnings and Precautions for Use
Endophthalmitis, retinal vasculitis and/or retinal occlusive vasculitis.
Intravitreal injections, including those with aflibercept, have been associated with endophthalmitis and more rarely, with retinal vasculitis and/or retinal occlusive vasculitis (see Section 4.8 Adverse Effects (Undesirable Effects)). Proper aseptic injection technique must always be used when administering Afqlir. Patients should be instructed to report any symptoms suggestive of endophthalmitis, retinal vasculitis or retinal occlusive vasculitis without delay and should be managed appropriately.
Retinal detachment.
Intravitreal injections, including those with aflibercept, have been associated with retinal detachment (see Section 4.8 Adverse Effects (Undesirable Effects)).
Increase in intraocular pressure.
Increases in intraocular pressure have been seen within 60 minutes of an intravitreal injection, including with aflibercept (see Section 4.8 Adverse Effects (Undesirable Effects)). Special precaution is needed in patients with poorly controlled glaucoma. In all cases both the intraocular pressure and the perfusion of the optic nerve head must therefore be monitored and managed appropriately.
Immunogenicity.
As this is a therapeutic protein, there is a potential for immunogenicity. Patients should be instructed to report any signs or symptoms of intraocular inflammation, e.g. pain, photophobia, or redness, which may be a clinical sign attributable to hypersensitivity.
Arterial thromboembolic events.
There is a potential risk of arterial thromboembolic events (ATEs) following intravitreal use of VEGF inhibitors (see Section 4.8 Adverse Effects (Undesirable Effects)). ATEs include vascular death (e.g. due to stroke or myocardial infarction), non-fatal strokes and non-fatal myocardial infarction.
The risk of stroke may be greater in patients with known risk factors including a history of stroke or transient ischaemic attack (TIA). Patients should be carefully evaluated by their doctor to assess whether the benefits of treatment outweigh the potential risks.
Bilateral treatment.
Bilateral treatment with Afqlir should be avoided. The safety and efficacy of bilateral treatment with aflibercept have not been systematically studied (see Section 5.1 Pharmacodynamic Properties, Clinical trials). If bilateral treatment is performed at the same time this could lead to an increased systemic exposure, which could increase the risk of systemic adverse events.
Retinal pigment epithelial tear.
Risk factors associated with the development of a retinal pigment epithelial tear after anti-VEGF therapy for wet AMD include a large and/or high pigment epithelial retinal detachment. When initiating anti-VEGF therapy, caution should be used in patients with these risk factors for retinal pigment epithelial tears.
Withholding treatment.
Treatment should be withheld in patients with rhegmatogenous retinal detachment or stage 3 or 4 macular holes.
In the event of a retinal break the dose should be withheld and treatment should not be resumed until the break is adequately repaired.
In the event of either a decrease in best-corrected visual acuity (BCVA) of ≥ 30 letters compared with the last assessment of visual acuity; or a subretinal haemorrhage involving the centre of the fovea or if the size of the haemorrhage is ≥ 50% of the total lesion area, the dose should be withheld and treatment should not be resumed earlier than the next scheduled treatment.
The dose should be withheld in the event of performed or planned intraocular surgery within the previous or next 28 days.
In patients presenting with clinical signs of irreversible ischaemic visual function loss, the treatment is not recommended.
Populations with limited data.
There is only limited experience with aflibercept treatment in diabetic patients with an HbA1c over 12% or with proliferative diabetic retinopathy or Type 1 diabetes. Aflibercept has not been studied in patients with active systemic infections or in patients with concurrent eye conditions such as retinal detachment or macular hole. There is also no experience of treatment with aflibercept in patients with uncontrolled hypertension. In myopic CNV there is no experience with aflibercept in the treatment of non-Asian patients, patients who have previously undergone treatment for myopic CNV, and patients with extrafoveal lesions.
This lack of information should be considered by the ophthalmologist when treating such patients.
Use in the elderly.
Available data do not suggest a need for a dose adjustment with aflibercept 2 mg in these patients (see Section 5.1 Pharmacodynamic Properties, Clinical trials). There is limited experience in patients with DME aged 75 years and older.
Paediatric use.
The safety and efficacy of aflibercept have not been studied in children or adolescents.
Effects on laboratory tests.
No relevant effects on laboratory tests are known.4.5 Interactions with Other Medicines and Other Forms of Interactions
No formal drug interaction studies have been performed with aflibercept.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Effects on male and female fertility were assessed as part of a 6-month study in monkeys with intravenous administration of aflibercept at doses ranging from 3 to 30 mg/kg every one to two weeks. Absent or irregular menses associated with alterations in female reproductive hormone levels and changes in sperm morphology and motility (considered consequential to male fertility) were observed at all dose levels. Based on Cmax and AUC for free aflibercept observed at the 3 mg/kg intravenous dose, the systemic exposures were approximately 4900-fold and 1500-fold higher, respectively, than the exposure observed in humans after an intravitreal dose of 2 mg. All changes were reversible.
(Category D)
There are limited data on the use of aflibercept in pregnant women. Women of childbearing potential have to use effective contraception during treatment and for at least 3 months after the last intravitreal injection of aflibercept 2 mg.
Afqlir should not be used during pregnancy unless the potential benefit outweighs the potential risk to the foetus. The treating ophthalmologist in consultation with the treating obstetrician need to consider the individual benefit-risk balance for each patient. This includes a consideration of timing of treatment, delaying treatment and other potential treatment options.
Studies in animals have shown reproductive toxicity, including a series of external, visceral, skeletal malformations, after systemic administration.
Aflibercept produced malformations and other fetal abnormalities in pregnant rabbits with intravenous administration (at 3 to 60 mg/kg once every 3 days during the period of organogenesis) and with subcutaneous administration (0.1 to 1 mg/kg on gestational days 1, 7, and 13). A No Observed Effect Level (NOEL) for adverse effects on embryofetal development was not established. At the lowest dose tested (0.1 mg/kg), the systemic exposures based on Cmax and cumulative AUC for free aflibercept were approximately 13- and 10-fold higher, respectively, when compared to corresponding values observed in humans after an intravitreal dose of 2 mg.
It is unknown whether aflibercept is excreted in human milk. A risk to the breast-fed child cannot be excluded. Afqlir is not recommended during breast-feeding. A decision must be made whether to discontinue breast-feeding or to abstain from Afqlir therapy.4.7 Effects on Ability to Drive and Use Machines
Patients may experience temporary visual disturbances after an intravitreal injection with aflibercept and the associated eye examinations (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients should not drive or use machinery until visual function has recovered sufficiently.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
A total of 3102 patients treated with aflibercept constituted the safety population in eight Phase III studies. Amongst those, 2501 patients were treated with the recommended dose of 2 mg.
Serious adverse reactions related to the injection procedure have occurred in less than 1 in 2400 intravitreal injections with aflibercept and included endophthalmitis, retinal detachment, cataract traumatic, cataract, vitreous detachment and intraocular pressure increased (see Section 4.4 Special Warnings and Precautions for Use).
The most frequently observed adverse reactions (in at least 5% of patients treated with aflibercept) were conjunctival haemorrhage (25.0%), visual acuity reduced (11.1%), eye pain (10.2%), cataract (7.6%), intraocular pressure increased (7.5%), vitreous detachment (7.4%), and vitreous floaters (6.9%).
In wet AMD, these adverse reactions occurred with a similar incidence in the ranibizumab treatment group.
Tabulated list of adverse reactions.
The safety data described in Table 1 include all adverse reactions (serious and non-serious) from eight Phase III studies with a reasonable possibility of causality to the injection procedure or medicinal product over the 96 weeks study duration for wet AMD, over 100 weeks for CRVO, over 100 weeks for DME, over 52 weeks for BRVO and over 48 weeks for myopic CNV.
The adverse reactions are listed by system organ class and frequency using the following convention: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000 patients). Within each frequency grouping, adverse drug reactions are presented in order of decreasing seriousness.
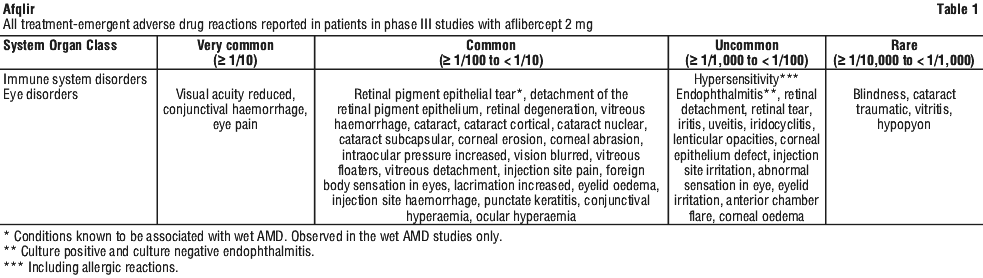
Post-marketing experience.
In addition, the following adverse reactions have also been reported during the post-marketing period of aflibercept 2 mg, for which a frequency could not be estimated.
Immune system disorders.
Hypersensitivity (including rash, pruritus, urticaria, and isolated cases of severe anaphylactic/anaphylactoid reactions).
Eye disorders.
Retinal vasculitis and retinal occlusive vasculitis, scleritis.
Description of selected adverse reactions.
In the wet AMD phase III studies, there was an increased incidence of conjunctival haemorrhage in patients receiving anti-thrombotic agents. This increased incidence was comparable between patients treated with ranibizumab and aflibercept.
Arterial thromboembolic events (ATEs) are adverse events potentially related to systemic VEGF inhibition. There is a theoretical risk of arterial thromboembolic events following intravitreal use of VEGF inhibitors.
ATEs, as defined by Antiplatelet Trialists' Collaboration (APTC) criteria, include nonfatal myocardial infarction, nonfatal stroke, or vascular death (including deaths of unknown cause). The incidence of adjudicated APTC ATEs in the VIEW 1 and VIEW 2 wet AMD studies during the 96 weeks study period was 3.3% (60 out of 1824) in the combined group of patients treated with aflibercept (2.4% in the aflibercept 2Q4 arm and 3.6% in the aflibercept 2Q8 arm), compared to 3.2% (19 out of 595) in patients treated with ranibizumab.
The incidence of adjudicated APTC ATEs in the CRVO studies (GALILEO and COPERNICUS) during the 76/100 weeks study duration was 0.6% (2 out of 317) in patients treated with at least one dose of aflibercept compared to 1.4% (2 out of 142) in the group of patients receiving only sham treatment.
The incidence of adjudicated APTC ATEs in the DME studies (VIVIDDME and VISTADME) during the 100 weeks study duration was 6.4% (37 out of 578) in the combined group of patients treated with aflibercept compared with 4.2% (12 out of 287) in the control group.
The incidence of APTC ATEs in the BRVO study (VIBRANT) during the 52 week study duration was 0% (0 out of 91) in patients treated with aflibercept compared with 2.2% (2 out of 92) in the control group.
The incidence of APTC ATEs in the myopic CNV study (MYRROR) during the 48 week study duration was 1.1% (1 out of 91) in the group of patients treated with aflibercept compared to 0% (0 out of 31) in the group of patients in the control group.
As with all therapeutic proteins, there is a potential for immunogenicity with aflibercept.
Comparability of Afqlir (aflibercept 2 mg) with Eylea (aflibercept 2 mg) in terms of safety.
The safety of Afqlir was assessed over 52 weeks in patients with wet AMD in study CSOK583A12301 (see Section 5.1 Pharmacodynamic Properties, Clinical trials). The mean number of study treatment injections was 7.6 in both the Afqlir (n=244) and Eylea (n=240) arms. The frequency and severity of ocular treatment-emergent adverse events were broadly comparable between Afqlir 2 mg and Eylea 2 mg.
Serious non-ocular treatment-emergent adverse events were reported in 14.2% and 11.3% of patients treated with Afqlir and Eylea, respectively. Arterial thromboembolic events, defined as nonfatal myocardial infarction, nonfatal stroke, or vascular death (including deaths of unknown cause), were reported in 2.9% and 0.8% of patients treated with Afqlir and Eylea, respectively.
Comparability of Afqlir (aflibercept 2 mg) with Eylea (aflibercept 2 mg) in terms of immunogenicity.
Immunogenicity was evaluated in 465 subjects in study CSOK583A12301. No clinically meaningful difference was found between Afqlir and Eylea in terms of the incidence of treatment-emergent anti-drug antibodies (ADAs).
Reporting of suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
Overdosing with increased injection volume may increase intraocular pressure.
Therefore, in case of overdosage intraocular pressure should be monitored and if deemed necessary by the treating ophthalmologist, adequate treatment should be initiated (see Section 4.2 Dose and Method of Administration, Method of administration).
For information on the management of overdose, contact the Poisons Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: ophthalmologicals/ antineovascularisation agents.
ATC code: S01LA05.
Mechanism of action.
Vascular endothelial growth factor-A (VEGF-A) and placental growth factor (PlGF) are members of the VEGF family of angiogenic factors that can act as potent mitogenic, chemotactic, and vascular permeability factors for endothelial cells. VEGF acts via two receptor tyrosine kinases, VEGFR-1 and VEGFR-2, present on the surface of endothelial cells. PlGF binds only to VEGFR-1, which is also present on the surface of leukocytes. Excessive activation of these receptors by VEGF-A can result in pathological neovascularisation and excessive vascular permeability. PlGF can synergise with VEGF-A in these processes, and is also known to promote leukocyte infiltration and vascular inflammation. A variety of ocular diseases is associated with pathologic neovascularisation and vascular leakage, and/or can result in thickening and oedema of the retina, which is thought to contribute to vision loss.
Aflibercept acts as a soluble decoy receptor that binds VEGF-A and PlGF with higher affinity than their natural receptors, and thereby can inhibit the binding and activation of these cognate VEGF receptors. The equilibrium dissociation constant (KD) for aflibercept binding to human VEGF-A165 is 0.5 picoM and to human VEGF-A121 is 0.36 picoM. The KD for binding to human PlGF-2 is 39 picoM.
Pharmacodynamic effects.
Neovascular (wet) age-related macular degeneration (wet AMD).
Wet AMD is characterised by pathological choroidal neovascularisation (CNV). Leakage of blood and fluid from CNV may cause retinal oedema and/or sub-/intra-retinal haemorrhage, resulting in loss of visual acuity.
In patients treated with aflibercept (one injection per month for three consecutive months, followed by one injection every 2 months), retinal thickness decreased soon after treatment initiation, and the mean CNV lesion size was reduced, consistent with the results seen with ranibizumab 0.5 mg every month.
In pivotal phase III clinical studies, VIEW 1 and VIEW 2, there were mean decreases in retinal thickness on time domain optical coherence tomography (OCT) at week 52: -130 and 129 microns for the aflibercept 2 mg every two months and ranibizumab 0.5 mg every month study groups, respectively, in VIEW 1; -149 and -139 microns for the aflibercept 2 mg every two months, and ranibizumab 0.5 mg every month study groups, respectively, in VIEW 2.
The reduction of CNV size and reduction in retinal thickness were generally maintained in the second year of the studies.
The supportive study, ALTAIR, enrolled Japanese patients with treatment naive wet AMD, using 3 initial monthly aflibercept 2 mg injections, followed by one injection after 2 months, and then continued with a treat-and-extend regimen with variable treatment intervals (2-week or 4-week adjustments) up to a maximum 16 week interval according to pre-specified criteria. At week 52, there were mean decreases in central retinal thickness (CRT) on spectral domain OCT of -134.4 and -126.1 microns for the 2-week adjustment group and the 4-week adjustment group, respectively. The proportion of patients without fluid on OCT at week 52 was 68.3% and 69.1% in the 2- and 4-week adjustment groups, respectively.
The reduction in retinal thickness was generally maintained in both treatment arms in the second year of the ALTAIR study.
Macular oedema following central retinal vein occlusion (CRVO).
In CRVO, retinal ischaemia occurs and signals the release of VEGF which in turn destabilises the tight junctions and promotes endothelial cell proliferation. Up-regulation of VEGF is associated with the breakdown of the blood retina barrier and this increased vascular permeability results in retinal oedema, stimulation of endothelial cell growth and neovascularisation.
In patients treated with aflibercept (one injection every month for six months), there was consistent, rapid and robust response in morphology (CRT as assessed by OCT). Improvements in mean CRT were maintained through week 24.
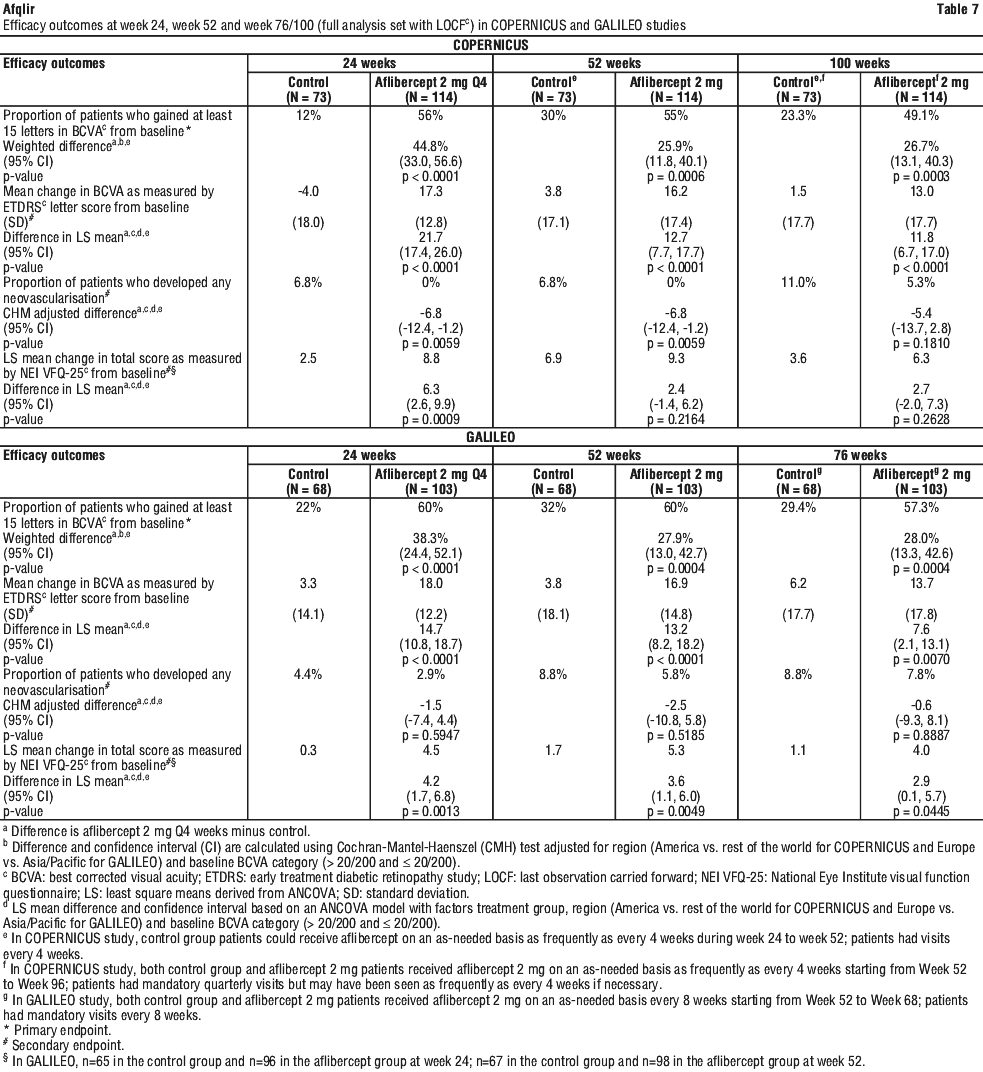
Retinal thickness on OCT at week 24 compared to baseline was a secondary efficacy endpoint in both the COPERNICUS and GALILEO studies. In both studies, the mean change in CRT from baseline to week 24 statistically significantly favoured aflibercept.
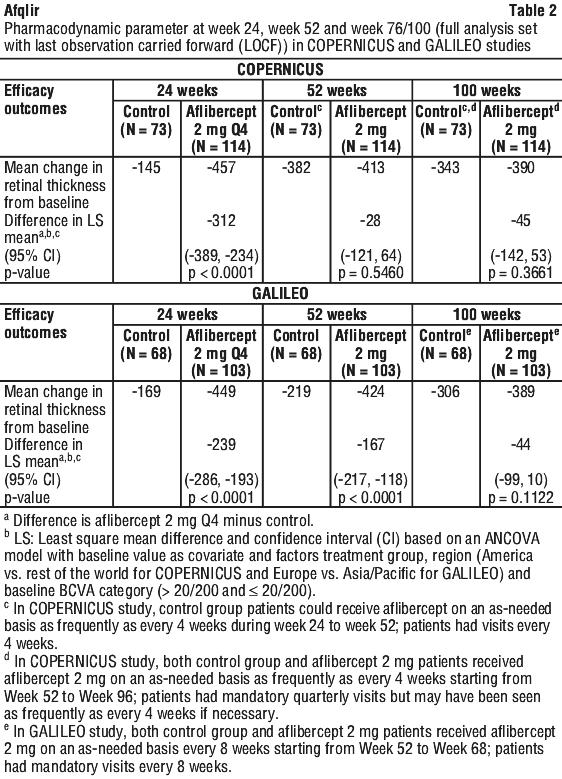
Macular oedema following branch retinal vein occlusion (BRVO).
In BRVO, retinal ischaemia occurs and signals the release of VEGF, which in turn destabilises the tight junctions and promotes endothelial cell proliferation. Up-regulation of VEGF is associated with the breakdown of the blood retina barrier and this increased vascular permeability results in retinal oedema, stimulation of endothelial cell growth and neovascularisation.
In patients treated with aflibercept (one injection every month for six months) in the VIBRANT study, there was consistent, rapid and robust response in retinal morphology (CRT as assessed by OCT). There was a statistically significant improvement in the aflibercept 2 mg group in comparison to the active control group treated with laser photocoagulation at week 24 (-280 microns vs. -128 microns). At week 24, the dosing interval was extended to every 2 months, and anatomic outcomes were maintained.
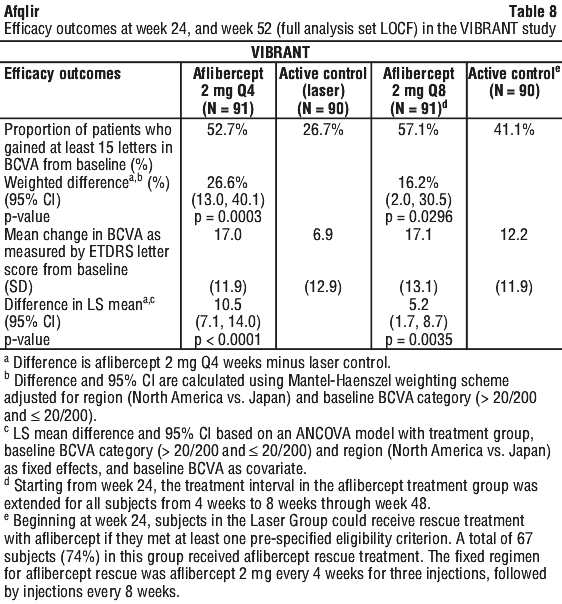
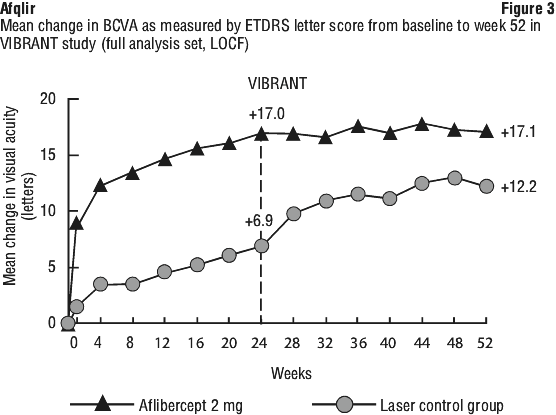
Retinal thickness on OCT at week 24 compared to baseline was a secondary efficacy variable in the VIBRANT study. This decrease from baseline was maintained to week 52, favouring aflibercept. See Table 3.
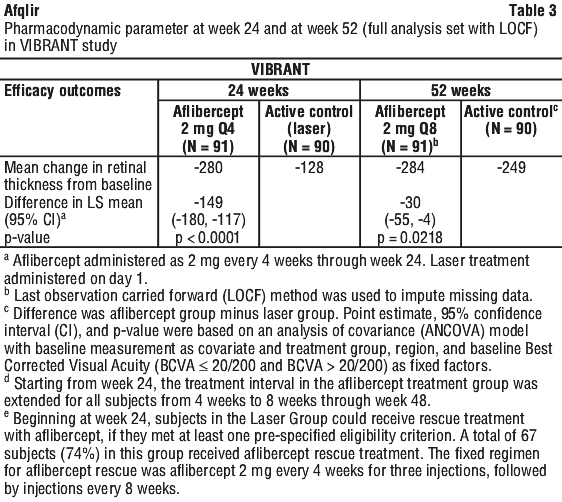
Diabetic macular oedema (DME).
Diabetic macular oedema is characterised by increased vasopermeability and damage to the retinal capillaries which may result in loss of visual acuity.
In patients treated with aflibercept, rapid and robust response in morphology (CRT) as assessed by OCT was seen soon after treatment initiation. The mean change in CRT from baseline to week 52 was statistically significant favouring aflibercept and was maintained through week 100. See Table 4.
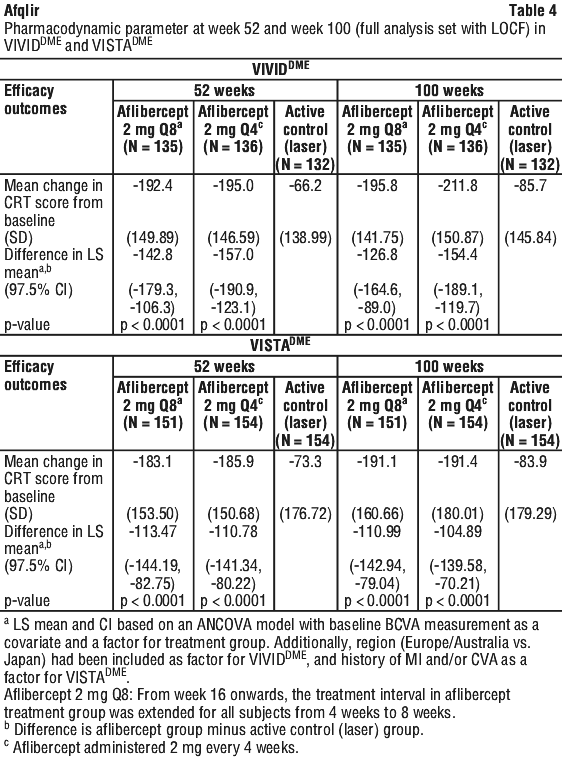 The VIOLET study compared three different dosing regimens of aflibercept 2 mg for treatment of DME. Following 5 consecutive monthly doses and treatment at fixed 8 week intervals for at least 1 year, patients continued treatment with aflibercept 2 mg according to one of the dosing regimens:
The VIOLET study compared three different dosing regimens of aflibercept 2 mg for treatment of DME. Following 5 consecutive monthly doses and treatment at fixed 8 week intervals for at least 1 year, patients continued treatment with aflibercept 2 mg according to one of the dosing regimens:
treat-and-extend (2TandE) where treatment intervals were maintained at a minimum of 8 weeks and gradually extended based on clinical and anatomical outcomes;
pro re nata (2PRN) where patients were observed every 4 weeks and injected when needed based on clinical and anatomical outcomes; and
dosed every 8 weeks (2Q8) for the second and third year of treatment.
At week 52 of the study, i.e. after at least two years of treatment, the mean changes in CRT from baseline were -2.1, 2.2 and -18.8 microns for 2TandE, 2PRN, and 2Q8 respectively. At week 100, i.e. after at least three years of treatment, the mean changes in CRT from baseline were 2.3, -13.9 and -15.5 microns, respectively (see Clinical trials).
Myopic choroidal neovascularisation (myopic CNV).
Myopic CNV is a frequent cause of vision loss in adults with pathologic myopia. Eyes with pathologic myopia are elongated, often excessively, and have, in addition, pathologic tissue alterations such as retinal pigment epithelial thinning and defects, lacquer cracks and Bruch's membrane ruptures, choroidal neovascularisation, subretinal haemorrhage and choroidal atrophy. As a consequence of ruptures of Bruch's membrane, myopic CNV develops as a wound healing mechanism and at the same time represents the most vision-threatening event in pathologic myopia.
In patients treated with aflibercept (one injection given at the start of therapy, additional injection given in case of disease persistence or recurrence) retinal thickness assessed by OCT decreased soon after treatment initiation and the mean CNV lesion size was reduced. The mean change in CRT from baseline to week 24 was statistically significant favouring aflibercept. See Table 5.
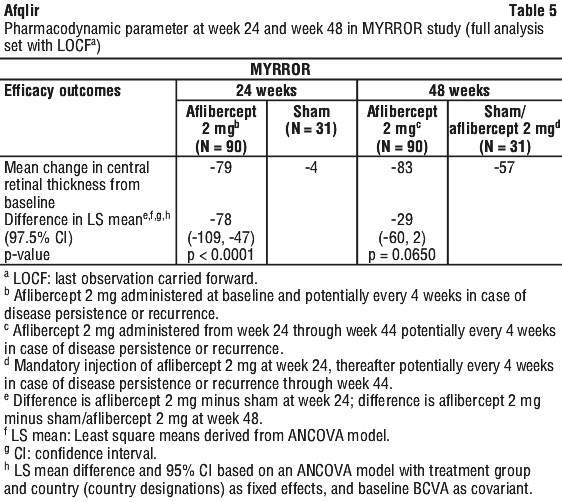
Clinical trials.
Neovascular (wet) age-related macular degeneration (wet AMD).
The safety and efficacy of aflibercept 2 mg were assessed in two pivotal phase III randomised, multi-centre, double-masked, active-controlled studies in patients with wet AMD. A total of 2412 patients were treated and evaluable for efficacy (1817 with aflibercept) in the two studies (VIEW 1 and VIEW 2). In each study, patients were randomly assigned in a 1:1:1:1 ratio to 1 of 4 dosing regimens:
1. Aflibercept administered at 2 mg every 8 weeks following 3 initial monthly doses (aflibercept 2Q8).
2. Aflibercept administered at 2 mg every 4 weeks (aflibercept 2Q4).
3. Aflibercept administered at 0.5 mg every 4 weeks (aflibercept 0.5Q4).
4. Ranibizumab administered at 0.5 mg every 4 weeks (ranibizumab 0.5Q4).
Patient ages ranged from 49 to 99 years with a mean of 76 years. Approximately 89% (1616/1817) of the patients randomised to treatment with aflibercept were 65 years of age or older and approximately 63% (1139/1817) were 75 years of age or older.
In the follow-up exploratory phase of the studies (i.e. from week 52 onwards to week 96), patients continued to receive the dosage strength to which they were initially randomised but on a modified dosing schedule. Injections were given as frequently as every 4 weeks, but no less frequently than every 12 weeks based upon pre-specified retreatment criteria guided by assessment of visual and/or anatomic outcomes. After the first year of the studies, 90% of patients originally treated with aflibercept 2Q8 received 6 doses or less and 72% received 4 doses or less among the patients completing the follow-up exploratory phase of the studies.
In both studies, the primary efficacy endpoint was the proportion of patients in the Per Protocol Set who maintained vision, defined as losing fewer than 15 letters of visual acuity at week 52 compared to baseline. The studies were intended to test for non-inferiority against ranibizumab 0.5 mg given every 4 weeks.
In the VIEW 1 study, at week 52, 95.1% of patients in the aflibercept 2Q8 treatment group maintained vision compared to 94.4% of patients in the ranibizumab 0.5Q4 group. Aflibercept treatment was shown to be non-inferior to the ranibizumab 0.5Q4 group.
In the VIEW 2 study, at week 52, 95.6% of patients in the aflibercept 2Q8 treatment group maintained vision compared to 94.4% of patients in the ranibizumab 0.5Q4 group. Aflibercept treatment was shown to be non-inferior to the ranibizumab 0.5Q4 group.
The VIEW 1 and VIEW 2 studies included four secondary efficacy endpoints: mean change in Best Corrected Visual Acuity (BCVA), proportion of patients who gained ≥ 15 letters, change in the total National Eye Institute Visual Function Questionnaire (NEI VFQ-25) score, and change in CNV area.
Detailed results from the combined analysis of both studies (primary* and secondary# endpoints) are shown in Table 6 and Figure 1.
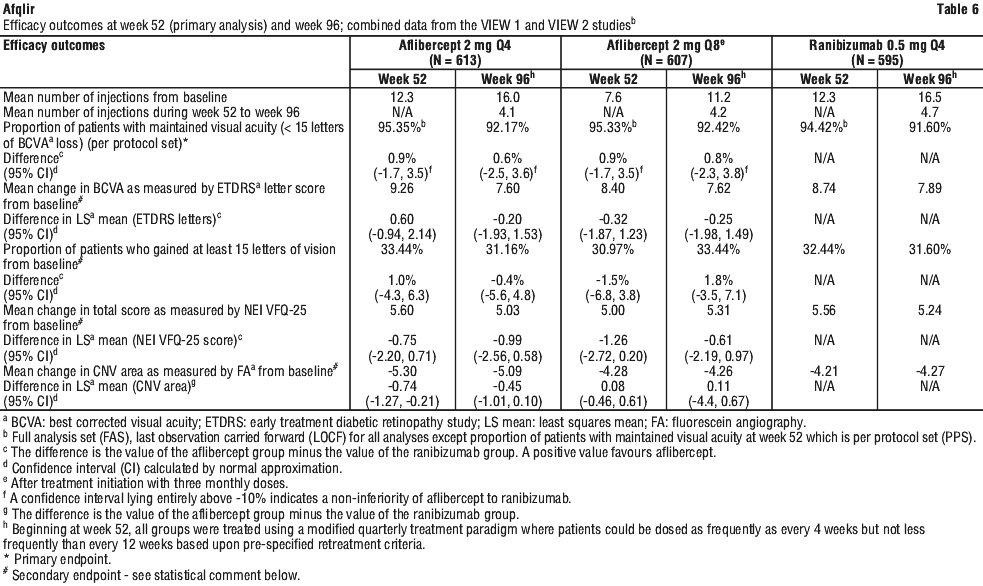
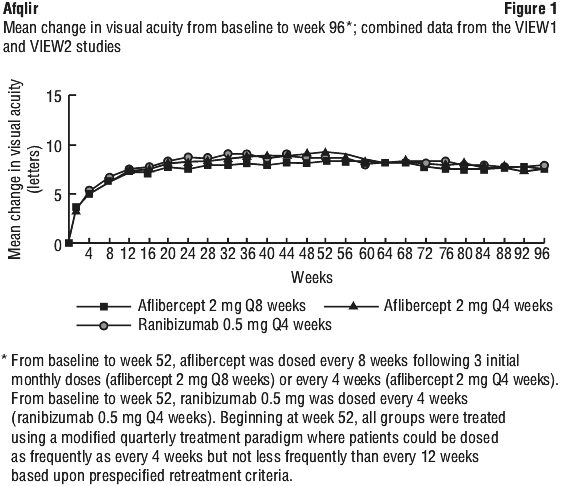 While there were small differences between aflibercept 2 mg and ranibizumab, no clinically relevant differences were seen between the treatment groups across all four secondary efficacy endpoints, based on the confidence intervals for the differences between aflibercept and ranibizumab. All statistical tests on secondary efficacy endpoints were considered to be exploratory in the combined analysis of both studies. All secondary endpoint analyses supported the comparability of the efficacy of all 3 aflibercept treatment schedules and ranibizumab.
While there were small differences between aflibercept 2 mg and ranibizumab, no clinically relevant differences were seen between the treatment groups across all four secondary efficacy endpoints, based on the confidence intervals for the differences between aflibercept and ranibizumab. All statistical tests on secondary efficacy endpoints were considered to be exploratory in the combined analysis of both studies. All secondary endpoint analyses supported the comparability of the efficacy of all 3 aflibercept treatment schedules and ranibizumab.
In combined data analysis of the VIEW 1 and VIEW 2 studies aflibercept demonstrated clinically meaningful changes from baseline in NEI VFQ-25 scores and subscales (near activities, distance activities, and vision-specific dependency). The magnitude of these changes was similar to that seen in published studies, which corresponded to a 15-letter gain in BCVA.
After the first year of the studies, efficacy was generally maintained through the last assessment at week 96. Over the 96 weeks period, patients in the aflibercept 2Q8 group received an average of 11.2 doses and patients in the ranibizumab group received an average of 16.5 doses.
Exploratory analyses of efficacy results in all evaluable subgroups (e.g. age, gender, race, baseline visual acuity, lesion type, lesion size) in each study and in the combined analysis were consistent with the results in the overall populations.
The supportive study, ALTAIR, is a 96 week Phase IV multicentre, randomised, open-label study in 247 Japanese patients with treatment naive wet AMD, designed to assess the efficacy and safety of aflibercept following two different adjustment intervals (2-weeks and 4-weeks) of a treat-and-extend dosing regimen.
All patients received 3 monthly doses of aflibercept 2 mg, followed by one injection after a further 2 month interval. At week 16, patients were randomised 1:1 into two treatment groups: 1) aflibercept treat-and-extend with 2-week adjustments and 2) aflibercept treat-and-extend with 4-week adjustments. Extension or shortening of the treatment interval was decided based on visual and/or anatomic criteria defined by protocol with a maximum treatment interval of 16 weeks for both groups.
The primary efficacy endpoint was mean change in BCVA from baseline to week 52. The secondary efficacy endpoints were the proportion of patients who did not lose ≥ 15 letters and the proportion of patients who gained at least 15 letters of BCVA from baseline to week 52.
At week 52, patients in the treat-and-extend arm with 2-week adjustments gained a mean of 9.0 letters from baseline as compared to 8.4 letters for those in the 4-week adjustment group [LS mean difference in letters (95% CI): -0.4 (-3.8,3.0), ANCOVA]. The proportion of patients who did not lose ≥ 15 letters in the two treatment arms was similar (96.7% in the 2-week and 95.9% in the 4-week adjustment groups). The proportion of patients who gained ≥ 15 letters at week 52 was 32.5% in the 2-week adjustment group and 30.9% in the 4-week adjustment group. The proportion of patients who extended their treatment interval to 12 weeks and beyond was 42.3% in the 2-week adjustment group and 49.6% in the 4-week adjustment group. Furthermore, in the 4-week adjustment group 40.7% of patients were extended to 16 week intervals. Ocular and systemic safety profiles were similar to the safety observed in the pivotal studies VIEW1 and VIEW2. There are no data directly comparing aflibercept administered in a treat-and extend dosing regimen with aflibercept administered every 8 weeks following 3 initial monthly doses during the first 12 months of treatment of wet AMD.
In the second year of the study, efficacy was generally maintained up to and including the last assessment at week 96, with a mean gain from baseline of 7.6 letters for the 2-week adjustment group and 6.1 letters for the 4-week adjustment group. The proportion of patients who extended their treatment interval to 12 weeks or beyond was 56.9% in the 2-week adjustment group and 60.2% in the 4-week adjustment group. At the last visit prior to week 96, 64.9% and 61.2% of patients in the 2-week and 4-week adjustment groups, respectively, had their next injection scheduled at an interval of 12 weeks or beyond.
Between week 16 and 96, 43.1% (n = 53) and 54.5% (n = 67) of the patients (2-week and 4-week adjustment groups respectively) were extended to a maximum treatment interval of 16 weeks at least once. Of these patients, 96.2% (n = 51 of 53) patients in the 2-week adjustment group and 77.6% (n = 52 of 67) patients in the 4-week adjustment group maintained a 16-week treatment interval until the end of the study. During the 96 week study period, 41.5% (n=51) and 46.3% (n=57) of patients in the 2-week and 4-week adjustment groups respectively had a final treatment interval of 16 weeks.
During the second year of treatment patients in both the 2-week and 4-week adjustment groups received an average of 3.6 and 3.7 injections. Over the 2-year treatment period patients received an average of 10.4 injections.
Macular oedema secondary to central retinal vein occlusion (CRVO).
The safety and efficacy of aflibercept were assessed in two randomised, multi-centre, double-masked, sham-controlled studies in patients with macular oedema secondary to CRVO. A total of 358 patients were treated and evaluable for efficacy (217 with aflibercept) in the two studies (COPERNICUS and GALILEO). In both studies, patients were randomly assigned in a 3:2 ratio to either 2 mg aflibercept administered every 4 weeks (2Q4) or the control group receiving sham injections every 4 weeks for a total of 6 injections.
After 6 monthly injections, patients received treatment only if they met pre-specified retreatment criteria, except for patients in the control group in the GALILEO study who continued to receive sham (control to control) until week 52. Starting from this time point, all patients were offered treatment if they met pre-specified criteria.
Patient ages ranged from 22 to 89 years with a mean of 64 years. Approximately 52% (112/217) of the patients randomised to treatment with aflibercept were 65 years of age or older and approximately 18% (38/217) were 75 years of age or older.
In both studies, the primary efficacy endpoint was the proportion of patients who gained at least 15 letters in BCVA at week 24 compared to baseline. The studies were designed to evaluate superiority against the control group (receiving sham injections).
Change in visual acuity at week 24 compared to baseline was an important secondary endpoint in both COPERNICUS and GALILEO studies.
The difference between treatment groups was statistically significant in favour of aflibercept in both studies, for the proportion of patients who gained at least 15 letters in BCVA and for mean change in visual acuity, at week 24 compared to baseline. In both pivotal studies, the maximal improvement in visual acuity was achieved at month 3 with subsequent stabilisation of the effect on visual acuity and central retinal thickness until month 6. The statistically significant difference was maintained through week 52. A difference was maintained through week 76/100.
Three other secondary endpoints were included in the studies: change in CRT, as assessed by OCT, at week 24 compared to baseline (see Section 5.1 Pharmacodynamic Properties, Pharmacodynamic effects); proportion of patients progressing to neovascularisation (anterior segment neovascularisation, neovascularisation of the optic disk, or neovascularisation of the retina elsewhere) at week 24; and change in the NEI VFQ25 total score at week 24 compared to baseline.
Detailed results from the analysis of both studies (primary* and secondary# endpoints) are shown in Table 2 (see Section 5.1 Pharmacodynamic Properties, Pharmacodynamic effects), Table 7 and Figure 2.
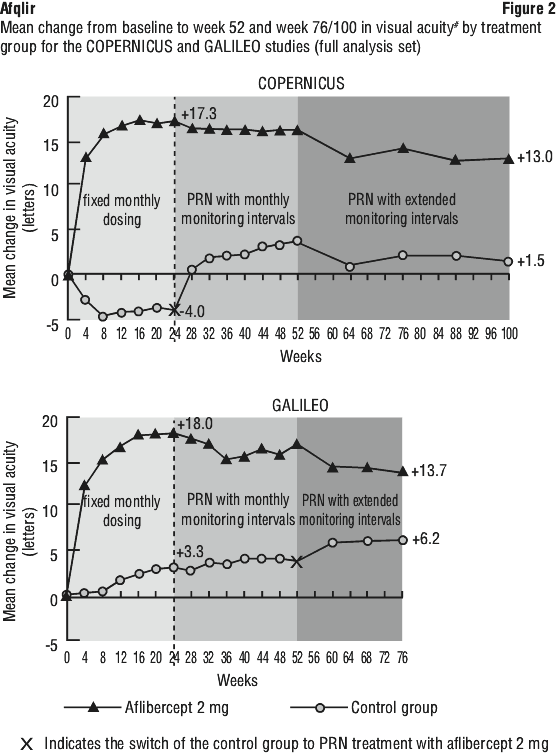 Exploratory analyses of efficacy results in all evaluable subgroups (e.g. age, gender, race, baseline visual acuity, retinal perfusion status, CRVO duration) in each study were in general consistent with the results in the overall populations.
Exploratory analyses of efficacy results in all evaluable subgroups (e.g. age, gender, race, baseline visual acuity, retinal perfusion status, CRVO duration) in each study were in general consistent with the results in the overall populations.
Macular oedema secondary to branch retinal vein occlusion (BRVO).
The safety and efficacy of aflibercept 2 mg were assessed in a randomised, multi-centre, double-masked, active-controlled study in patients with macular oedema secondary to BRVO, which included Hemi-Retinal Vein Occlusion. A total of 181 patients were treated and evaluable for efficacy (91 with aflibercept) in the VIBRANT study. In the study, patients were randomly assigned in a 1:1 ratio to either 2 mg aflibercept administered every 4 weeks, with a total of 6 injections, or laser photocoagulation administered at baseline (laser control group).
Patients in the laser control group could receive additional laser photocoagulation (called "rescue laser treatment") beginning at week 12, if at least one pre-specified rescue treatment criterion was met. The minimum interval between laser photocoagulation treatments was 12 weeks. After week 24, patients in the aflibercept group received 2 mg every 8 weeks through week 48, and patients in the control group could receive treatment with aflibercept 2 mg, if at least one pre-specified rescue criterion was met. Aflibercept rescue treatment consisted of a fixed regimen with 2 mg aflibercept administered every 4 weeks for 3 injections, followed by intravitreal injections every 8 weeks through week 48.
Patient ages ranged from 42 to 94 years with a mean of 65 years. Approximately 58% (53/91) of the patients randomised to treatment with aflibercept were 65 years of age or older, and approximately 23% (21/91) were 75 years of age or older.
In the VIBRANT study, the primary efficacy endpoint was the proportion of patients who gained at least 15 letters in BCVA at Week 24 compared to baseline. At Week 24, the aflibercept group was superior to laser control for the primary endpoint.
Change in visual acuity at week 24 compared to baseline was a secondary efficacy variable in the VIBRANT study. The difference between treatment groups was statistically significant in favour of aflibercept. The course of visual improvement was rapid and maximal improvement was achieved at week 12, with subsequent stabilisation of the effect on visual acuity and central retinal thickness until week 24 and subsequent maintenance of the effect until week 52.
In the laser group 67 patients (74%) received rescue treatment with aflibercept beginning at week 24. In this treatment group, visual acuity improved by about 5 letters from week 24 to 52.
Detailed results from the analysis of the VIBRANT study are shown in Table 8 and Figure 3.
 The proportion of retinal perfused patients in the aflibercept group at baseline was 60.4% (n = 55). At week 24, this proportion increased to 80.2% (n = 65) and was sustained at week 52 (77.9%, n = 67). The proportion of perfused patients that started on grid laser photocoagulation was 68.9% (n = 62) at baseline. Perfusion at the week 24 primary endpoint in the laser group was 67.1% (n = 55). Patients in the laser group were eligible for rescue treatment with aflibercept beginning at week 24 according to pre-specified criteria. At week 52, 78.0% (n = 64) were perfused at this time.
The proportion of retinal perfused patients in the aflibercept group at baseline was 60.4% (n = 55). At week 24, this proportion increased to 80.2% (n = 65) and was sustained at week 52 (77.9%, n = 67). The proportion of perfused patients that started on grid laser photocoagulation was 68.9% (n = 62) at baseline. Perfusion at the week 24 primary endpoint in the laser group was 67.1% (n = 55). Patients in the laser group were eligible for rescue treatment with aflibercept beginning at week 24 according to pre-specified criteria. At week 52, 78.0% (n = 64) were perfused at this time.
The beneficial effect of aflibercept treatment on visual function was similar in the baseline groups with perfused and non-perfused patients.
Treatment effects in evaluable subgroups (e.g. age, gender, and baseline retinal perfusion status) in the study were in general consistent with the results in the overall populations.
Diabetic macular oedema (DME) (aflibercept 2 mg).
The safety and efficacy of aflibercept 2 mg were assessed in two randomised, multi-centre, double-masked, active-controlled studies in patients with DME. A total of 862 randomised and treated patients were evaluable for efficacy. Of those, 576 were randomised to the aflibercept groups in two studies (VIVIDDME and VISTADME). In each study, patients were randomly assigned in a 1:1:1 ratio to 1 of 3 dosing regimens:
1. Aflibercept administered at 2 mg every 8 weeks following 5 initial monthly injections (aflibercept 2Q8);
2. Aflibercept administered at 2 mg every 4 weeks (aflibercept 2Q4); and
3. macular laser photocoagulation (active control).
Beginning at week 24, patients meeting a pre-specified threshold of vision loss were eligible to receive additional treatment: patients in the aflibercept groups could receive laser and patients in the laser group could receive aflibercept.
Patient ages ranged from 23 to 87 years with a mean of 63 years. Approximately 47% (268/576) of the patients randomised to treatment with aflibercept were 65 years of age or older, and approximately 9% (52/576) were 75 years of age or older. Efficacy and safety outcomes were consistent with the outcomes of the overall population.
In both studies, the primary efficacy endpoint was the mean change from baseline in BCVA at Week 52 as measured by ETDRS letter score. Both aflibercept 2Q8 and aflibercept 2Q4 groups were shown to have efficacy that was statistically significantly superior to the laser control group. This benefit was maintained through week 100.
Detailed results from the analysis of the VIVIDDME and VISTADME studies are shown in Table 9 and Figure 4.
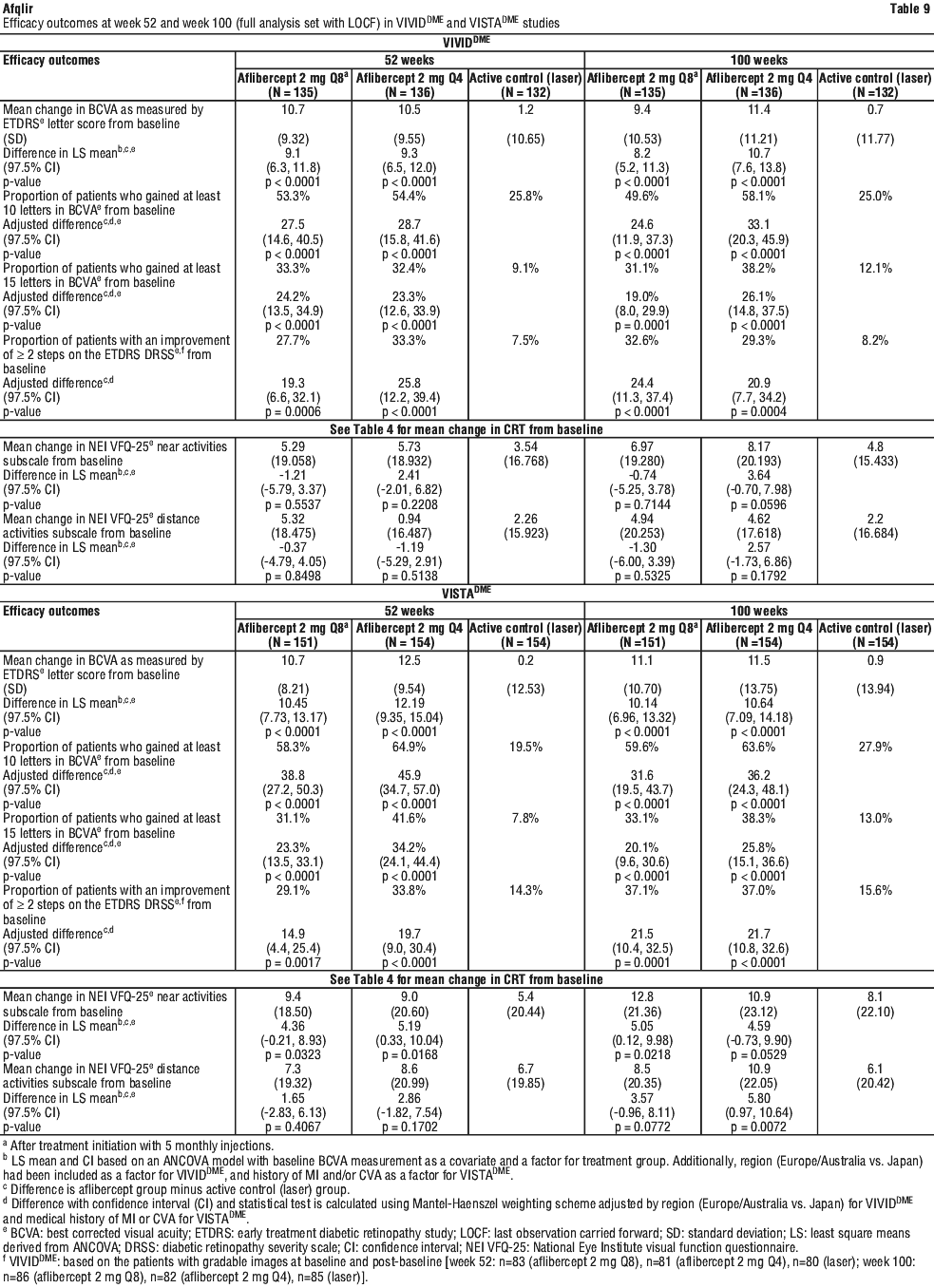
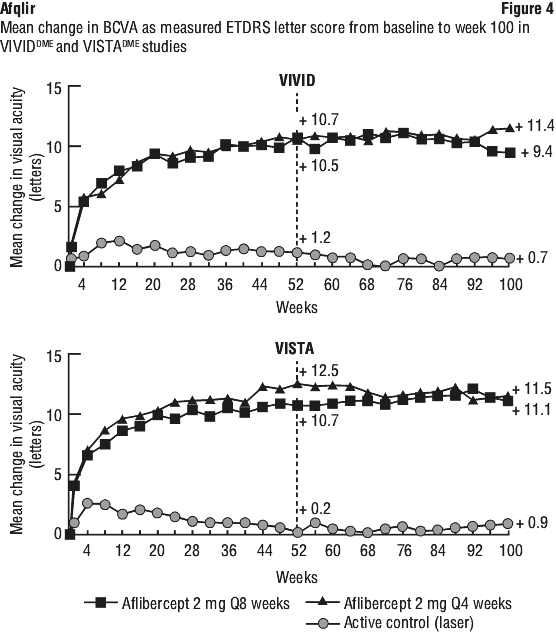 At week 52, 33.3% and 33.8% of 2Q4 patients, 27.7% and 29.1% of 2Q8 patients, and 7.5% and 14.3% of laser control patients in the VIVIDDME and VISTADME studies, respectively experienced an improvement in the severity of diabetic retinopathy, as measured by a ≥ 2 step improvement in the diabetic retinopathy severity scale (DRSS). This improvement was maintained through week 100 (see Table 9).
At week 52, 33.3% and 33.8% of 2Q4 patients, 27.7% and 29.1% of 2Q8 patients, and 7.5% and 14.3% of laser control patients in the VIVIDDME and VISTADME studies, respectively experienced an improvement in the severity of diabetic retinopathy, as measured by a ≥ 2 step improvement in the diabetic retinopathy severity scale (DRSS). This improvement was maintained through week 100 (see Table 9).
Treatment effects in evaluable subgroups (e.g. age, gender, race, baseline HbA1c, baseline visual acuity, prior anti-VEGF therapy) in each study and in the combined analysis were generally consistent with the results in the overall populations.
In the VIVIDDME and VISTADME studies, 36 (8.9%) and 197 (42.9%) patients received prior anti-VEGF therapy, respectively, with a 3-month or longer washout period. Treatment effects in the subgroup of patients who had previously been treated with a VEGF inhibitor prior to study participation were similar to those seen in patients who were VEGF inhibitor naive prior to study participation.
Patients with bilateral disease were eligible to receive anti-VEGF treatment in their fellow eye. In the VISTADME study, 217 (70.7%) of aflibercept patients received bilateral aflibercept injections until week 100; in the VIVIDDME study, 97 (35.8%) of aflibercept patients received a different anti-VEGF treatment in their fellow eye until week 100.
An independent comparative trial (DRCR.net Protocol T) utilised a flexible dosing regimen based on strict OCT and vision re-treatment criteria. In the aflibercept treatment group (n = 224) at week 52, this treatment regimen resulted in patients receiving a mean of 9.2 injections and mean gain of 13.3 letters, which was similar to the aflibercept 2Q8 group in VIVIDDME and VISTADME. (Mean number of injections: 8.7 and 8.4. Mean vision acquity improvement 10.7 letters). 42% of patients gained at least 15 letters in vision from baseline which also comparable to VIVIDDME and VISTADME (33.3% and 31.1% respectively). Safety outcomes demonstrated that overall incidence of ocular and non-ocular adverse events (including ATEs) were comparable across all treatment groups in each of the studies and between the studies.
A propensity score matching methodology (PSM) analysis compared the flexible aflibercept treatment group in Protocol T with the combined 2Q8 treatment groups in VIVID and VISTA.
This PSM identified, subsets of 179 matched patients from pooled VIVIDDME and VISTADME (utilising a fixed aflibercept dosing regimen) and Protocol T (utilising a flexible dosing regimen based on strict OCT and vision re-treatment criteria).
The PSM analysis showed that mean change in BCVA from baseline at week 52 was 10.9 letters in the 2 mg aflibercept 2Q8 fixed dosing regimen (VIVIDDME and VISTADME) and 13.7 letters in the 2 mg aflibercept flexible dosing regimen (Protocol T).
VIOLET was a 100-week multicentre, randomised, open-label, active controlled study in 463 patients with DME. Patients were randomised in a 1:1:1 ratio to three regimens of aflibercept 2 mg for treatment of DME after at least one year of treatment at fixed intervals, where treatment was initiated with 5 consecutive monthly doses followed by dosing every 2 months. The study evaluated non-inferiority of:
Aflibercept 2 mg dosed according to a treat-and-extend regimen (2TandE) where treatment intervals were maintained at a minimum of 8 weeks and gradually extended based on clinical and anatomical outcomes. The increments and decrements for the treatment intervals were at the investigator's discretion; increments of 2 weeks were recommended in the study; and
Aflibercept 2 mg dosed as needed (2PRN) where patients were observed every 4 weeks and injected when needed based on clinical and anatomical outcomes, compared to aflibercept 2 mg dosed every 8 weeks (2Q8).
The primary efficacy endpoint (change in BCVA from baseline to week 52) was 0.5 ± 6.7 letters in the 2TandE group and 1.7 ± 6.8 letters in the 2PRN group compared to 0.4 ± 6.7 letters in the 2Q8 group, achieving statistical non-inferiority (NI) (p < 0.0001 for both comparisons; NI margin 4 letters). The changes in BCVA from baseline to week 100 were consistent with the week 52 results: -0.1 ± 9.1 letters in the 2TandE group and 1.8 ± 9.0 letters in the 2PRN group compared to 0.1 ± 7.2 letters in the 2Q8 group. The mean number of injections over 100 weeks were 10.0, 11.5 and 12.3 for 2TandE, 2PRN and 2Q8, respectively.
Ocular and systemic safety profiles in all 3 treatment groups were similar to those observed in the pivotal studies VIVID and VISTA.
Myopic choroidal neovascularisation (myopic CNV).
The safety and efficacy of aflibercept 2 mg were assessed in a randomised, multi-centre, double-masked, sham-controlled study (MYRROR) in patients with myopic CNV. A total of 121 patients were treated and evaluable for efficacy (90 with aflibercept). Patients were randomly assigned in a 3:1 ratio to either 2 mg aflibercept administered once at study start (with additional injections given in the case of disease persistence or reoccurrence) or sham injections. In total 6 injections was possible until the week 24 primary endpoint assessment in the study.
After the first 6 months, patients initially randomised to sham were eligible to receive the first dose of aflibercept at week 24. Following this, patients in this former sham arm and also patients in the arm initially randomised to active treatment continued to be eligible for additional injections in case of disease persistence or recurrence.
Patient ages ranged from 27 to 83 years with a mean of 58 years. Approximately 36% (33/91) of the patients randomised to treatment with aflibercept were 65 years of age or older, and approximately 10% (9/91) were 75 years of age or older.
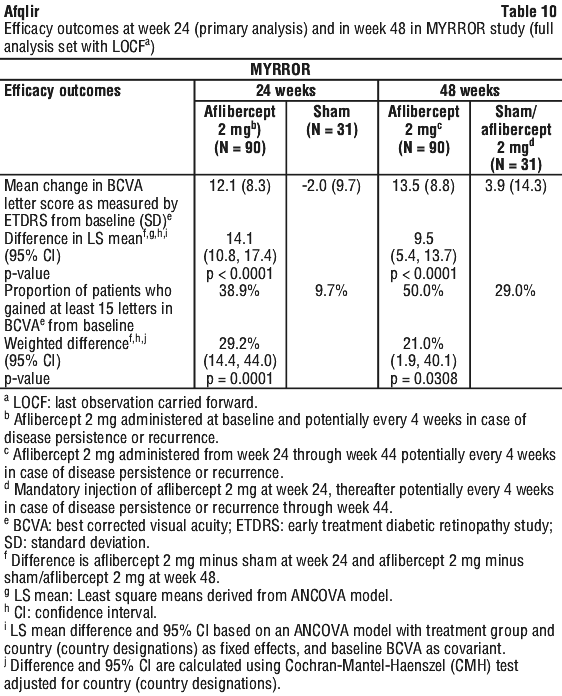
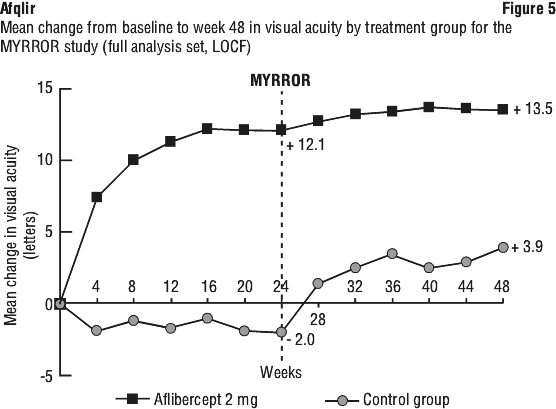
The primary efficacy endpoint was the change in visual acuity at week 24 compared to baseline. The confirmatory secondary efficacy endpoint was the proportion of patients who gained at least 15 letters in BCVA at week 24 compared to baseline.
The difference between treatment groups was statistically significant in favour of aflibercept for the primary and confirmatory secondary efficacy endpoints at week 24. Differences for both endpoints were maintained through week 48.
Detailed results from the analyses are shown in Table 10 and Figure 5.
 Treatment effects in all evaluable subgroups were in general, consistent with the results in the overall populations.
Treatment effects in all evaluable subgroups were in general, consistent with the results in the overall populations.
Comparative non-clinical pharmacology and toxicology.
Pharmacodynamic comparability between Afqlir and Eylea was demonstrated in in vitro studies.
Pharmacodynamic comparability of Afqlir (aflibercept 2 mg) with Eylea (aflibercept 2 mg).
No clinical comparative pharmacodynamic studies have been performed with Afqlir.
Comparability of Afqlir (aflibercept 2 mg) with Eylea (aflibercept 2 mg) in terms of efficacy.
Study CSOK583A12301 was an international, multicentre, randomised, double-masked, 2-arm parallel study in subjects with wet AMD, with a total duration of 52 weeks. The eligible subject population comprised male and female subjects who were 50 years of age or older, anti-VEGF treatment naive for both eyes, and diagnosed with active CNV secondary to AMD in the study eye.
A total of 485 subjects were randomised 1:1 to receive either Afqlir or Eylea, and 484 subjects were treated. 461 subjects were included in the Per-protocol Set (PPS) for analysis of the primary efficacy endpoint. Only 1 eye was selected as the study eye. Subjects received a single intravitreal injection of Afqlir 2 mg or Eylea 2 mg in the study eye every 4 weeks at 3 consecutive visits (Baseline, Week 4, and Week 8), and thereafter every 8 weeks at Weeks 16, 24, 32, 40, and 48.
The overall mean age in the PPS was 76 years (range 53 to 94), and 59% of subjects were 75 years or older. 56% of subjects were female and 89% were white. At baseline, the overall median time since diagnosis of nAMD in the PPS was 12 days, and less than 30 days had passed since diagnosis in 74% of patients. The overall mean BCVA score at baseline, as measured using ETDRS charts, was 59.7 letters. At baseline, 81% and 75% of subjects in the PPS had the occult lesion type in the Afqlir arm and the Eylea arm, respectively.
Primary efficacy was assessed at Week 8 after subjects have received two injections of either Afqlir 2 mg or Eylea 2 mg. At Week 8 the difference between Afqlir and Eylea in the LS mean changes in BCVA score from baseline was -0.3 letters for the PPS. The 95% CI (-1.8, 1.3) was contained within the prespecified interval [-3.5, +3.5]. Similar efficacy in terms of change in BCVA score from baseline was concluded. See Table 11.

5.2 Pharmacokinetic Properties
Aflibercept 2 mg is administered directly into the vitreous to exert local effects in the eye.
Absorption/ distribution.
Aflibercept is slowly absorbed from the eye into the systemic circulation after intravitreal administration and is predominantly observed in the systemic circulation as an inactive, stable complex with VEGF; however only free aflibercept is able to bind endogenous VEGF.
In a pharmacokinetic sub-study with frequent sampling in patients with wet AMD, maximum plasma concentrations of free aflibercept (systemic Cmax) were low, with a mean of approximately 0.02 microgram/mL (range 0 to 0.054) within 1 to 3 days after 2 mg intravitreal injection, and were undetectable two weeks following dosage in almost all patients. Aflibercept does not accumulate in the plasma when administered intravitreally every 4 weeks.
These pharmacokinetic results were consistent in pharmacokinetic sub-studies in patients with CRVO, BRVO, DME or myopic CNV, with mean Cmax of free aflibercept in plasma in the range of 0.03 to 0.05 microgram/mL and individual values not exceeding 0.14 microgram/mL. Thereafter, plasma concentrations of free aflibercept declined to values below or close to the lower limit of quantitation generally within one week; undetectable concentrations were reached before the next administration after 4 weeks in all patients. See Table 12.
 The mean maximum plasma concentration of free aflibercept is approximately 50 to 500 times below the aflibercept concentration required to inhibit the biologic activity of systemic VEGF by 50% in animal models. It is estimated that after intravitreal administration of 2 mg to patients, the mean maximum plasma concentration of free aflibercept is more than 100-fold lower than the concentration of aflibercept required to half-maximally bind systemic VEGF. Therefore, systemic pharmacodynamic effects are unlikely.
The mean maximum plasma concentration of free aflibercept is approximately 50 to 500 times below the aflibercept concentration required to inhibit the biologic activity of systemic VEGF by 50% in animal models. It is estimated that after intravitreal administration of 2 mg to patients, the mean maximum plasma concentration of free aflibercept is more than 100-fold lower than the concentration of aflibercept required to half-maximally bind systemic VEGF. Therefore, systemic pharmacodynamic effects are unlikely.
Metabolism.
As aflibercept is a protein-based therapeutic, no metabolism studies have been conducted.
Excretion.
Free aflibercept binds VEGF to form a stable, inert complex. As with other large proteins, both free and bound aflibercept are expected to be cleared by proteolytic catabolism.
Pharmacokinetic comparability of Afqlir (aflibercept 2 mg) with Eylea (aflibercept 2 mg).
A dedicated study to demonstrate pharmacokinetic comparability was not conducted due to the low systemic exposure of aflibercept after intravitreal injection.
5.3 Preclinical Safety Data
Genotoxicity.
No studies have been conducted on the mutagenic or clastogenic potential of aflibercept. As a large protein molecule, aflibercept is not expected to interact directly with DNA or other chromosomal material.
Carcinogenicity.
No studies have been conducted on the carcinogenic potential of aflibercept.6 Pharmaceutical Particulars
6.1 List of Excipients
Histidine, histidine hydrochloride monohydrate, trehalose dihydrate, polysorbate 20, hydrochloric acid, sodium hydroxide, water for injections.
6.2 Incompatibilities
Afqlir must not be mixed with other medicinal products.
6.3 Shelf Life
The expiry date can be found on the packaging. In Australia, information on the shelf life can be found on the public summary of the ARTG.
6.4 Special Precautions for Storage
Store at 2°C to 8°C (Refrigerate. Do not freeze). Protect from light.
Keep the vial in its carton in order to protect from light.
Keep the pre-filled syringe in its blister pack and carton in order to protect from light.
Prior to usage, the unopened vial or pre-filled syringe blister pack of Afqlir may be stored outside the refrigerator below 30°C for up to 14 days. Store in original carton and do not open vial or sealed blister pack until time of use. After opening the vial or blister pack, proceed under aseptic conditions.
6.5 Nature and Contents of Container
Afqlir is supplied in a single-use vial or pre-filled syringe.
Vial.
Each carton includes a type I glass vial containing approximately 240 microL of solution, which provides a usable amount to deliver a single dose of 50 microL containing 2 mg aflibercept. The vial is sealed with an elastomeric rubber stopper, and is provided with an 18 G filter needle.
Pre-filled syringe.
Each carton includes a sealed blister pack with a sterile pre-filled type I glass syringe, containing approximately 165 microL of solution, which provides a usable amount to deliver a single dose of 50 microL containing 2 mg aflibercept. The syringe is sealed with an elastomeric plunger stopper and an elastomeric tip cap that is part of a closure system with Luer lock adaptor. The syringe has a pre-attached plunger rod and a finger grip.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
The secondary and tertiary structures of aflibercept as well as the amino acid structure are shown.

 Chemical names: Vascular endothelial growth factor receptor type VEGFR-1 (synthetic human immunoglobulin domain 2 fragment) fusion protein with vascular endothelial growth factor receptor type VEGFR-2 (synthetic human immunoglobulin domain 3 fragment) fusion protein with immunoglobulin G1 (synthetic Fc fragment), dimer des-432-lysine[human vascular endothelial growth factor receptor 1-(103-204) peptide (containing Ig-like C2-type 2 domain) fusion protein with human vascular endothelial growth factor receptor 2-(206-308)-peptide (containing Ig-like C2-type 3 domain fragment) fusion protein with human immunoglobulin G1-(227 C-terminal residues)-peptide (Fc fragment)], (211-211':214-214')-bisdisulfide dimer.
Chemical names: Vascular endothelial growth factor receptor type VEGFR-1 (synthetic human immunoglobulin domain 2 fragment) fusion protein with vascular endothelial growth factor receptor type VEGFR-2 (synthetic human immunoglobulin domain 3 fragment) fusion protein with immunoglobulin G1 (synthetic Fc fragment), dimer des-432-lysine[human vascular endothelial growth factor receptor 1-(103-204) peptide (containing Ig-like C2-type 2 domain) fusion protein with human vascular endothelial growth factor receptor 2-(206-308)-peptide (containing Ig-like C2-type 3 domain fragment) fusion protein with human immunoglobulin G1-(227 C-terminal residues)-peptide (Fc fragment)], (211-211':214-214')-bisdisulfide dimer.
Molecular weight: 97 kDa (protein molecular weight), 115 kDa (total molecular weight).
CAS number.
862111-32-8.7 Medicine Schedule (Poisons Standard)
S4 - Prescription Only Medicine.
Summary Table of Changes
