What is in this leaflet
This leaflet answers some common questions about ALPROLIX. It does not contain all the available information. It does not use the place of talking to your doctor or pharmacist.
This leaflet was last updated on the date at the end of this leaflet.
Speak to your pharmacist or doctor to obtain the most up to date information on this medicine.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking ALPROLIX against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What ALPROLIX is used for
ALPROLIX is used for the management of haemophilia B (congenital factor IX deficiency). ALPROLIX is used to:
- control and prevent bleeding episodes
- routinely prevent and reduce the frequency of bleeding episodes
- reduce bleeding before, during, and after surgery.
People with haemophilia B lack sufficient factor IX to control bleeding. ALPROLIX works by replacing factor IX to enable blood to clot.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed it for another reason.
This medicine is not addictive.
It is available only with a doctor's prescription.
Do not give this medicine to anyone else. It may harm them, even if their symptoms are the same as yours.
Before you use ALPROLIX
When you must not use it
Do not use ALPROLIX if you have an allergy to:
- ALPROLIX or other factor IX replacement factors
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
If any of these signs occur, stop using ALPROLIX and see your doctor immediately.
Do not use this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
Do not use ALPROLIX if the medicine is cloudy, contains particles or is discoloured. It should be clear to slightly opalescent and colourless.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to use it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you are pregnant or intend to become pregnant. There is no information on the use of ALPROLIX during pregnancy. Your doctor will discuss the risks and benefits of taking it if you are pregnant.
Tell your doctor if you are breast-feeding or planning to breast-feed. It is not known whether ALPROLIX passes into breast milk. Your doctor will discuss the risks and benefits of taking it if you are breast-feeding.
If you have not told your doctor about any of the above, tell them before you start using ALPROLIX.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop. Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to use ALPROLIX
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
How much to use
Your doctor will decide how much ALPROLIX you use. This will depend on your individual need for replacement factor IX therapy. Your doctor may change the dose you use during your treatment.
Do not stop using ALPROLIX or change the dosage, without checking with your doctor unless you have an allergic reaction.
How to use it
ALPROLIX is given by slow injection directly into your veins.
ALPROLIX is provided as a powder and sterile sodium chloride solution 0.325% (diluent) which need to be mixed together before use.
It is important to not shake ALPROLIX when mixing it. Shaking can damage this medicine.
Mix the ALPROLIX powder with the diluent provided only when you are ready to use it. If you mix the powder and diluent and are interrupted, you can keep the mixed product for a maximum of 6 hours when stored at room temperature (below 30°C).
Do not put it in the freezer.
Always inspect ALPROLIX before use and after it has been mixed. The medicine should be clear to slightly opalescent and colourless.
Do not inject if the solution is discoloured or cloudy or contains particles.
Refer to the leaflet in the pack for step-by-step instructions about how to prepare and inject ALPROLIX.
Use in one patient on one occasion only. Dispose of all unused solution, empty vials, and used needles and syringes into a sharps bin.
Talk to your doctor or pharmacist, or telephone 1800 852 289 in Australia or 0800 852 289 in NZ, if you have any questions about how to use ALPROLIX.
How long to use it
Continue taking your medicine for as long as your doctor tells you.
This medicine helps to control your condition, but does not cure it.
If you forget to use it
Use your dose of ALPROLIX as soon as you remember, and resume your normal dosing schedule.
Do not use a double dose to make up for the dose that you missed. This may increase the chance of you getting an unwanted side effect.
If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering to use your medicine, ask your pharmacist for some hints.
If you use too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (in Australia telephone 13 11 26, in New Zealand telephone 0800 764 766) for advice, or go to Emergency at the nearest hospital, if you think that you or anyone else may have used too much ALPROLIX. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are using ALPROLIX
Things you must do
Tell your doctor immediately if bleeding is not controlled after using ALPROLIX.
If you become pregnant while on treatment with ALPROLIX, immediately tell your doctor.
Always talk to your doctor or pharmacist before taking any other medicine while you are using ALPROLIX.
Do not use more than the recommended dose.
Tell any other doctors, dentists and pharmacists who treat you that you are using this medicine.
If you are about to have any blood tests, tell your doctor that you are using ALPROLIX.
Keep all of your doctor's appointments so that your progress can be checked. Your doctor may do some blood tests before you start your treatment and from time to time during your treatment to monitor your progress.
Things you must not do
Do not use ALPROLIX to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they appear to have the same condition as you.
Do not stop using your medicine or change the dosage without checking with your doctor.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking ALPROLIX.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- headache
- tingling or numbness in your mouth (paraesthesia)
- breath odour
- fatigue
- dizziness
- taste disturbance or loss of taste (dysguesia)
- pain at site of infusion
- low blood pressure (symptoms include dizziness or feeling lightheaded)
- fast or irregular heartbeats, also called palpitations
- pain in your side with blood in your urine (obstructive uropathy).
The above list includes the more common side effects of your medicine. If any of these persist or worsen, talk to your doctor.
If any of the following happen, tell your doctor immediately or go to Emergency at your nearest hospital:
- swelling of your face, lips, tongue or other parts of the body, rash or hives
- shortness of breath, wheezing, difficulty breathing, chest pain or discomfort.
The above list includes very serious side effects. You may need urgent medical attention or hospitalisation. These side effects are very rare.
ALPROLIX may increase the risk of formation of abnormal blood clots in your body if you have risk factors for developing blood clots.
Your body can make antibodies called "inhibitors" against ALPROLIX, which may stop ALPROLIX from working properly
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
After using ALPROLIX
Storage
Keep your ALPROLIX in the pack until it is time to use it. This medicine should be protected from light.
Keep ALPROLIX in the refrigerator at 2°C to 8°C. If necessary, you can keep ALPROLIX out of the refrigerator for a single 6 month period. If out of the refrigerator, store the sealed carton in a cool dry place where the temperature stays below 30°C. The date that the product is removed from the refrigerator should be recorded on the carton.
Do not use any ALPROLIX that has been out of the refrigerator for more than 6 months (refer to Disposal below).
Once reconstituted, you can keep ALPROLIX at room temperature (below 30°C) for up to 6 hours. Protect the product from direct sunlight.
Do not store ALPROLIX or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Do not freeze ALPROLIX. Do not place in the freezer or freezing compartment of a refrigerator.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop using this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
ALPROLIX comes as a white to off-white powder to cake in a glass vial. Each pack contains:
- 1 vial of sterile ALPROLIX powder
- 1 pre-filled syringe of diluent
- 1 sterile vial adapter reconstitution device.
ALPROLIX is available in 6 strengths: 250 IU, 500 IU, 1000 IU, 2000 IU, 3000 IU and 4000IU.
Ingredients
ALPROLIX contains eftrenonacog alfa as the active ingredient.
Other ingredients:
- sucrose
- sodium chloride
- histidine
- mannitol
- polysorbate 20.
Further information
You can obtain more information from your doctor, pharmacist or your Haemophilia Treatment Centre, or by telephoning 1800 207 753 in Australia or 0800 852 289 in New Zealand.
Sponsor
ALPROLIX is supplied in Australia by:
sanofi-aventis pty ltd
12-24 Talavera Road
Macquarie Park NSW 2113
Freecall No: 1800 818 806
Email: [email protected]
ALPROLIX is supplied in New Zealand by:
Sanofi-aventis New Zealand limited
Level 8, 56 Cawley Street
Ellerslie, Auckland
New Zealand Free Call: 0800 283 684
Email: [email protected]
This leaflet was prepared in May 2020.
ALPROLIX 250 IU - AUST R 209227
ALPROLIX 500 IU - AUST R 209223
ALPROLIX 1000 IU - AUST R 209224
ALPROLIX 2000 IU - AUST R 209225
ALPROLIX 3000 IU - AUST R 209226
ALPROLIX 4000 IU - AUST R 315497
ALPROLIX® is a registered trademark of Bioverativ Therapeutics Inc.
Published by MIMS July 2020
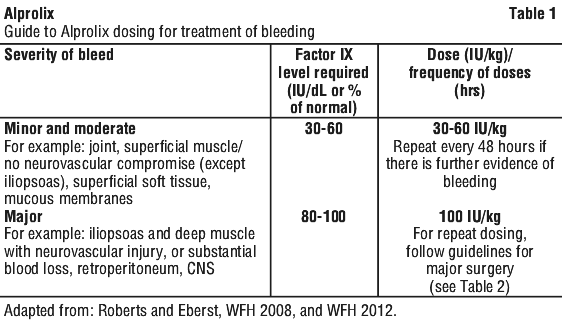 Subsequent dosage and duration of treatment depends on the individual clinical response, pharmacokinetic profile, the severity of the factor IX deficiency, and the location and extent of bleeding.
Subsequent dosage and duration of treatment depends on the individual clinical response, pharmacokinetic profile, the severity of the factor IX deficiency, and the location and extent of bleeding.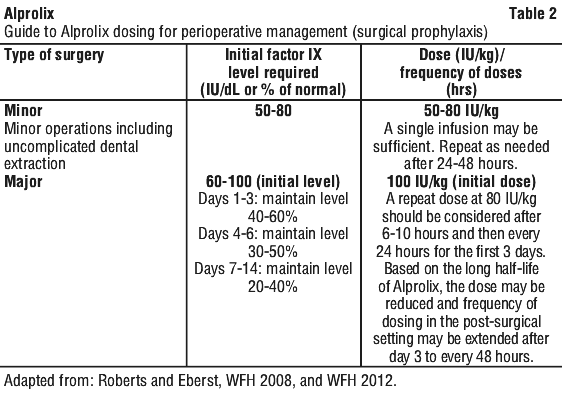 Higher doses or more frequent dosing may be needed in patients less than 12 years of age.
Higher doses or more frequent dosing may be needed in patients less than 12 years of age.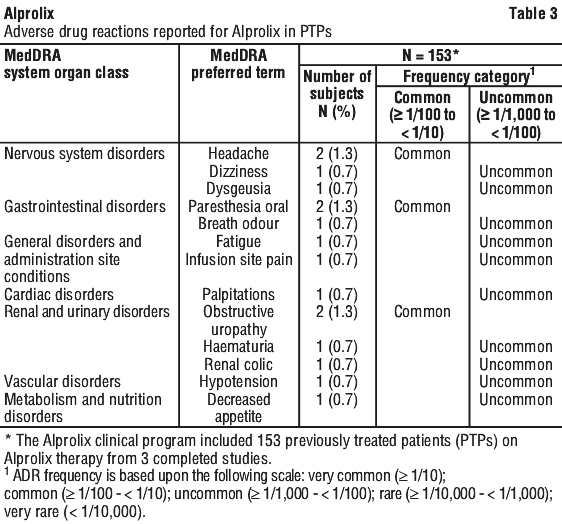
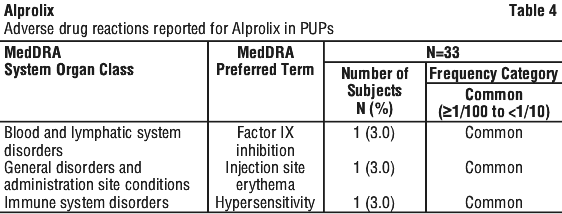 Both events of factor IX inhibition and hypersensitivity shown in Table 4 occurred in a single previously untreated subject while on Alprolix, after 10 Eds. This subject discontinued Alprolix due to the adverse drug reactions.
Both events of factor IX inhibition and hypersensitivity shown in Table 4 occurred in a single previously untreated subject while on Alprolix, after 10 Eds. This subject discontinued Alprolix due to the adverse drug reactions.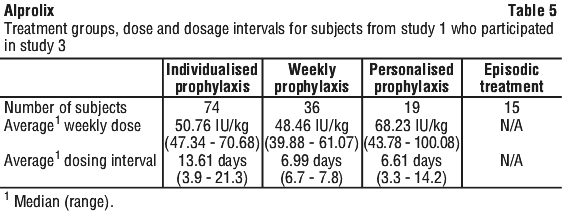 The majority of subjects from Study 2 stayed on their treatment regimen throughout the extension study, with 3 subjects (11.1%) switching treatment regimens once or twice during the study. From the start of Study 2 to the end of Study 3, subjects had a median of 150 (range: 16.9 to 251.1) cumulative weeks of treatment.
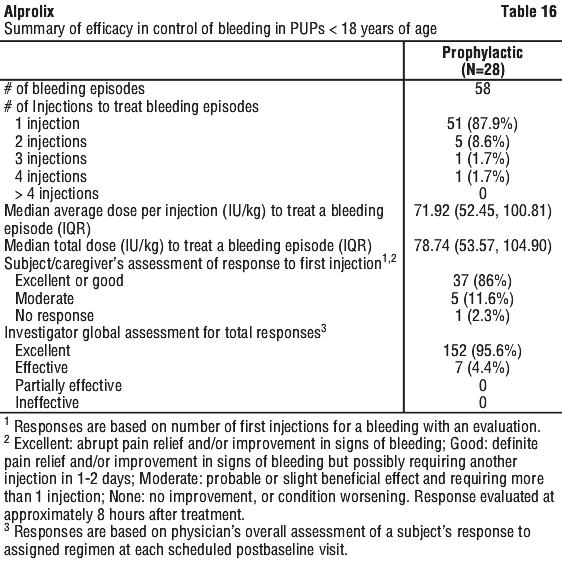
The majority of subjects from Study 2 stayed on their treatment regimen throughout the extension study, with 3 subjects (11.1%) switching treatment regimens once or twice during the study. From the start of Study 2 to the end of Study 3, subjects had a median of 150 (range: 16.9 to 251.1) cumulative weeks of treatment.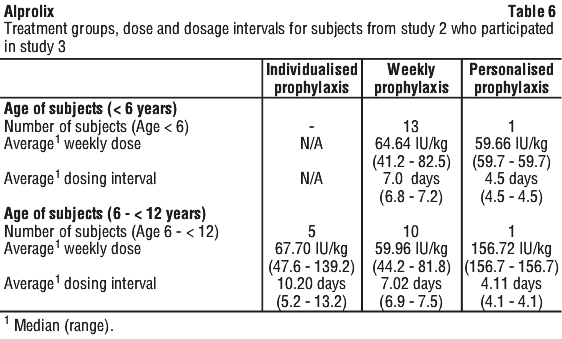 Study 4 enrolled 33 previously untreated patients (PUPs) < 18 years old with haemophilia B (≤ 2% endogenous FIX activity). The primary endpoint was the occurrence of inhibitor development. At enrolment, the median age was 0.6 years (range: 0.08-2 years), 78.8% of subjects were less than 1 year old, and the median weight was 9 kg (range: 4.6-17 kg). The overall median number of weeks on treatment was 83.01 (range: 6.7-226.7 weeks), and the overall median number of EDs was 76 days (range: 1-137 days) per subject. Subjects could be treated episodically (optional), until a prophylactic regimen was initiated.
Study 4 enrolled 33 previously untreated patients (PUPs) < 18 years old with haemophilia B (≤ 2% endogenous FIX activity). The primary endpoint was the occurrence of inhibitor development. At enrolment, the median age was 0.6 years (range: 0.08-2 years), 78.8% of subjects were less than 1 year old, and the median weight was 9 kg (range: 4.6-17 kg). The overall median number of weeks on treatment was 83.01 (range: 6.7-226.7 weeks), and the overall median number of EDs was 76 days (range: 1-137 days) per subject. Subjects could be treated episodically (optional), until a prophylactic regimen was initiated.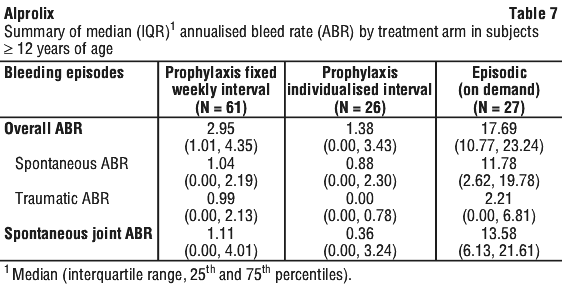
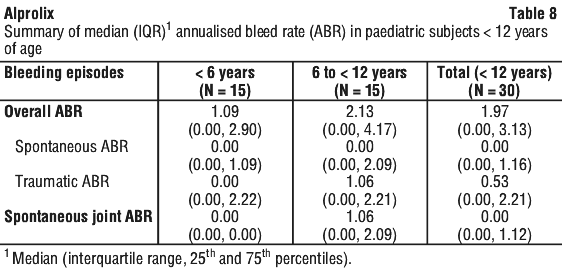
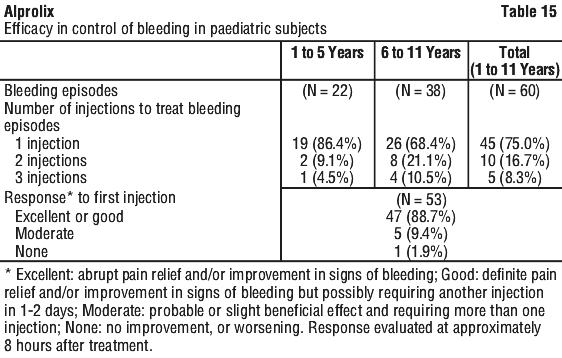
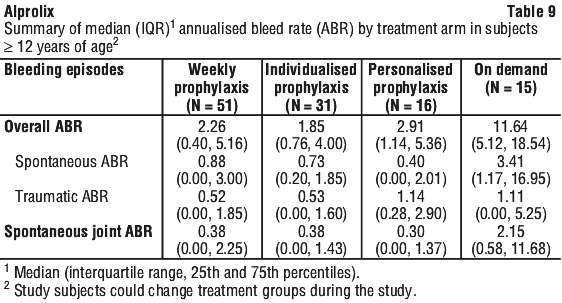 The overall median annualised bleeding rates in paediatric subjects < 12 years of age are presented in Table 10. The median duration of treatment on Study 2 was 55 weeks (range: 7.9-177.0) in subjects < 5 and 175.7 weeks (range: 47.0-201.1) in subjects 6 to < 12.
The overall median annualised bleeding rates in paediatric subjects < 12 years of age are presented in Table 10. The median duration of treatment on Study 2 was 55 weeks (range: 7.9-177.0) in subjects < 5 and 175.7 weeks (range: 47.0-201.1) in subjects 6 to < 12.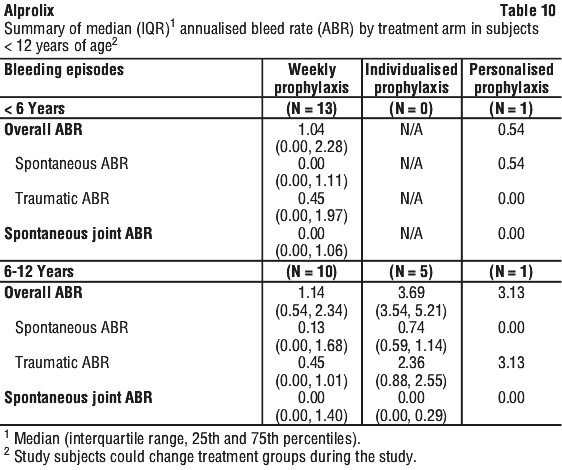
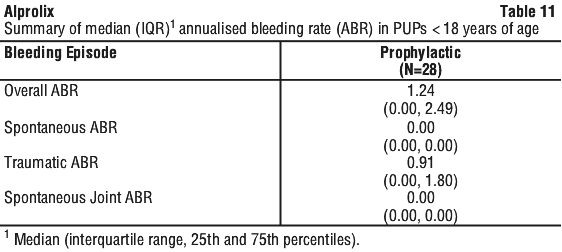

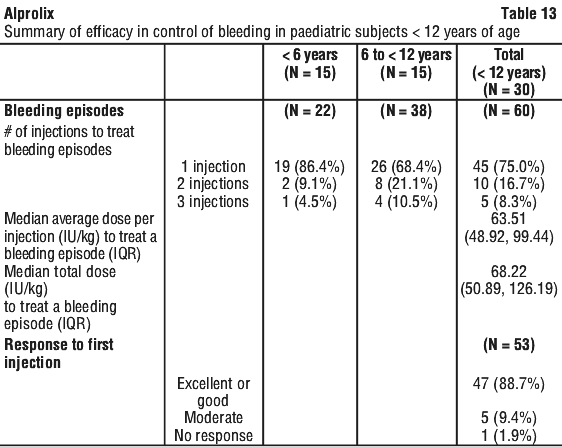

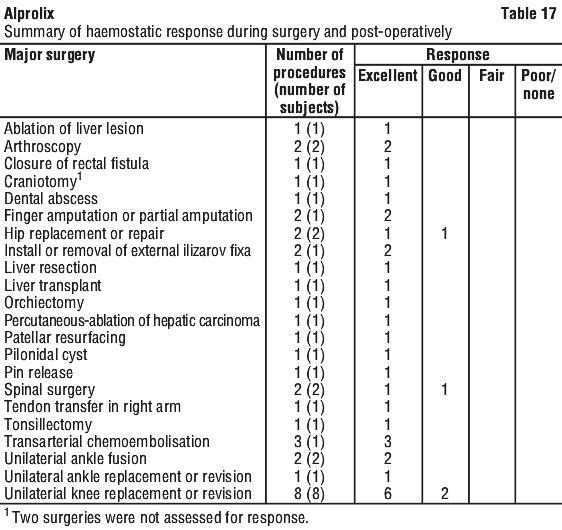
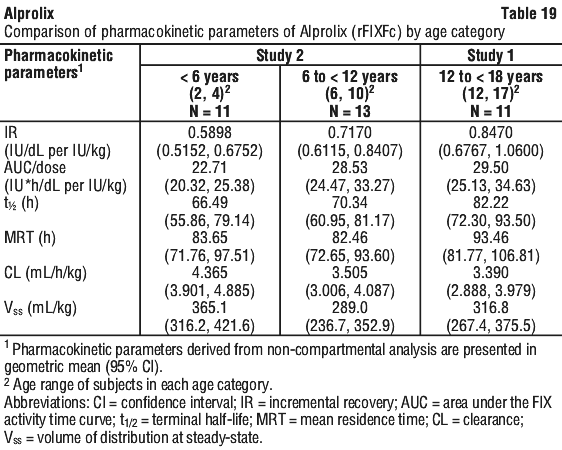
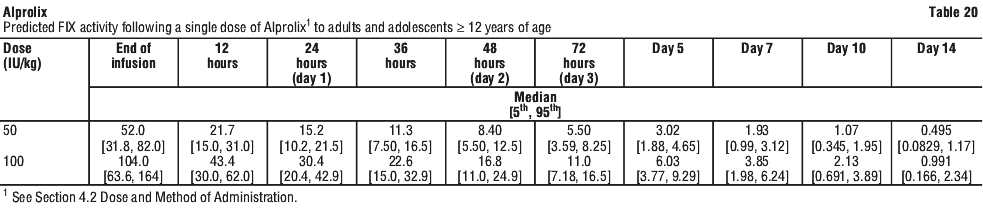
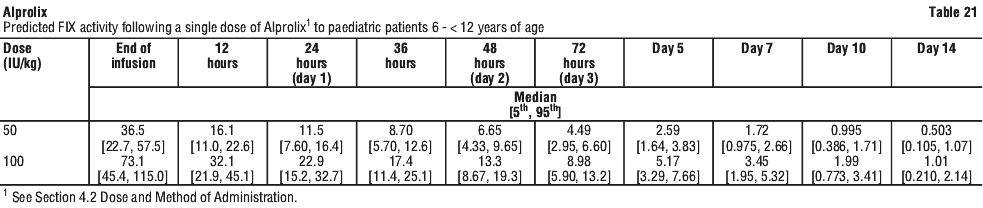
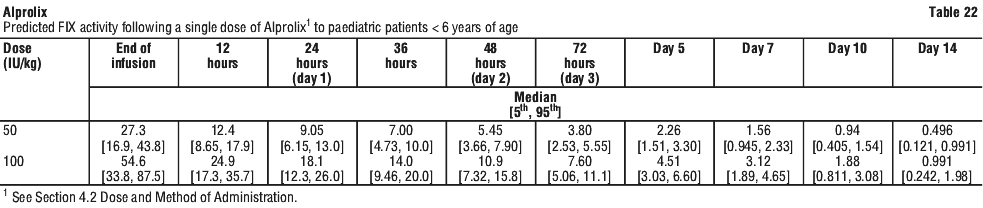 Measured FIX activity in 14 subjects undergoing surgical procedures in a clinical study was consistent with the values predicted by the population PK model. A sample perioperative dosing regimen to achieve target FIX levels for adults and adolescents, as simulated by this model, is shown in Table 23.
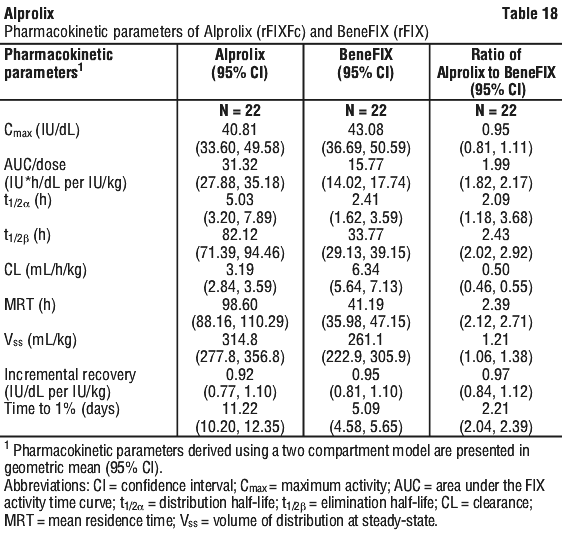
Measured FIX activity in 14 subjects undergoing surgical procedures in a clinical study was consistent with the values predicted by the population PK model. A sample perioperative dosing regimen to achieve target FIX levels for adults and adolescents, as simulated by this model, is shown in Table 23.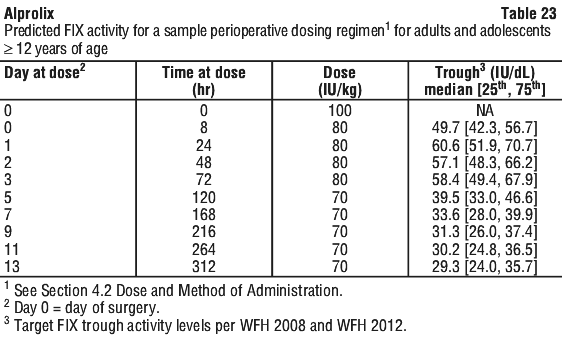 Alprolix has been evaluated in 161 male haemophilia B patients from 2 to 76 years of age (see Section 4.4 Special Warnings and Precautions for Use, Use in the elderly) and weighing between 12.5 to 186.7 kg. Body weight has an impact on the pharmacokinetics of Alprolix. Age does not provide any further impact beyond that provided by body weight.
Alprolix has been evaluated in 161 male haemophilia B patients from 2 to 76 years of age (see Section 4.4 Special Warnings and Precautions for Use, Use in the elderly) and weighing between 12.5 to 186.7 kg. Body weight has an impact on the pharmacokinetics of Alprolix. Age does not provide any further impact beyond that provided by body weight.