

What is in this leaflet
This leaflet answers some common questions about Alunbrig.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
The information in this leaflet was last updated on the date listed on the final page. More recent information on the medicine may be available. You should ensure that you speak to your pharmacist or doctor to obtain the most up to date information on this medicine. You can also download the most up to date leaflet from https://www.ebs.tga.gov.au.
Those updates may contain important information about the medicine and its use of which you should be aware.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking this medicine against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Alunbrig is used for
Alunbrig contains the active ingredient brigatinib.
Alunbrig belongs to a group of medicines called anti-neoplastic (or anti-cancer) agents which are used to treat cancer.
Alunbrig is used to treat adults with a type of lung cancer called non-small cell lung cancer. It is used if your cancer:
- is ALK-positive – this means your cancer cells have a fault in a gene called anaplastic lymphoma kinase (ALK)
- is advanced or has spread to another part of your body (metastatic)
- has previously been treated with a medicine called crizotinib, which is causing too many side effects or has stopped working.
Alunbrig prevents the activity of the ALK protein. This protein is known to be involved in the growth and spread of cancer cells.
Alunbrig may slow down or stop the growth of your cancer. It may also help to shrink your cancer.
Ask your doctor if you have any questions about why it has been prescribed for you. Your doctor may have prescribed it for another purpose.
This medicine is not addictive.
This is available only with a doctor's prescription.
Before you take Alunbrig
When you must not take it
Do not take Alunbrig if you have an allergy to:
- any medicine containing brigatinib
- any of the ingredients listed at the end of this leaflet
Some symptoms of an allergic reaction may include shortness of breath, wheezing or difficulty breathing, swelling of the face, lips, tongue or other parts of the body.
Do not take this medicine if you are pregnant. It may affect your developing baby if you take it during pregnancy.
Do not breastfeed if you are taking this medicine.
Do not give Alunbrig to children under the age of 18 years. Safety and effectiveness in children have not been established.
Do not take it after the expiry date printed on the pack or if the packaging is damaged or shows signs of tampering. If it has expired or is damaged return it to your pharmacist for disposal.
Before you start to take it
Tell your doctor if you have any allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- lung or breathing problems
- high blood pressure
- a slow heartbeat
- problems with your vision
- sore or painful muscles
- problems with your pancreas
- high blood sugar
Tell your doctor if you are pregnant or intend to become pregnant. Alunbrig may be harmful to an unborn baby when taken by a pregnant woman. You should not take Alunbrig while you are pregnant.
If you are a woman who could become pregnant, use highly effective contraception (birth control) during treatment, and for at least 4 months after taking the last tablet.
If you are the partner of a woman who could become pregnant, use highly effective contraception during treatment, and for at least 3 months after taking the last tablet.
Talk to your doctor about the right methods of contraception for you and your partner.
Tell your doctor if you are breastfeeding or plan to breastfeed. It is not known whether brigatinib passes into breast milk. It is not recommended that you breastfeed while taking Alunbrig.
If you have not told your doctor about any of the above, tell them before you take Alunbrig.
Taking other medicines
Tell your doctor if you are taking any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food store.
Some medicines and Alunbrig may interfere with each other. These include:
- ketoconazole, itraconazole, voriconazole: medicines to treat fungal infections
- indinavir, nelfinavir, ritonavir, saquinavir: medicines to treat HIV infection
- clarithromycin, telithromycin, troleandomycin: medicines to treat bacterial infections
- St. John's wort: a herbal product used to treat depression
- carbamazepine: a medicine to treat epilepsy, euphoric/ depressive episodes and certain pain conditions
- phenobarbital, phenytoin: medicines to treat epilepsy
- rifabutin, rifampicin: medicines to treat tuberculosis or certain other infections
- digoxin: a medicine to treat heart weakness
- dabigatran: a medicine to inhibit blood clotting
- colchicine: a medicine to treat gout attacks
- pravastatin, rosuvastatin: medicines to lower elevated cholesterol levels
- methotrexate: a medicine to treat severe joint inflammation, cancer and the skin disease psoriasis
- sulfasalazine: a medicine to treat severe bowel and rheumatic joint inflammation
- efavirenz, etravirine: medicines to treat HIV infection
- modafinil: a medicine to treat narcolepsy
- bosentan: a medicine to treat pulmonary hypertension
These medicines may be affected by Alunbrig, or may affect how well it works. You may need to use different amounts of your medicine, or take different medicines.
Your doctor or pharmacist has more information on medicines to be careful with or to avoid while taking Alunbrig.
How to take Alunbrig
Follow all directions given to you by your doctor carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions, ask your doctor or pharmacist for help.
How much to take
Your doctor will tell you how many tablets you need to take each day. This may depend on your condition, whether you are taking any other medicines and whether you experience side effects.
The usual dose of Alunbrig is one 90 mg tablet once daily for the first 7 treatment days; thereafter, one 180 mg tablet once daily.
How to take it
Swallow the tablets whole with a full glass of water. Do not crush or dissolve the tablets.
Alunbrig tablets can be taken with or without food.
When to take it
Take Alunbrig once a day at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
How long to take it
The duration of treatment with Alunbrig varies, depending on the nature of your illness and your individual response to treatment.
Continue taking Alunbrig until your doctor tells you to stop.
If you forget to take it
If you forget to take a dose, or if you vomit after taking a dose, skip the dose you missed and take your next dose when you are meant to.
Do not take a double dose to make up for the dose that you missed. This may increase the chance of getting an unwanted side effect.
If you are not sure what to do, ask your doctor.
If you have trouble remembering when to take your medicine, ask your pharmacist for hints.
If you take too much (overdose)
Immediately telephone your doctor, or the Poisons Information Centre (telephone Australia 13 11 26), or go to Accident and Emergency at your nearest hospital, if you think you or anyone else may have taken too much Alunbrig. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
If you are not sure what to do, ask your doctor.
While you are taking Alunbrig
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking Alunbrig.
Tell any other doctors, dentists and pharmacists who treat you that you are taking Alunbrig.
Use highly effective contraception to prevent pregnancy while you are being treated with Alunbrig. Women must avoid pregnancy during treatment with Alunbrig and for at least 4 months after taking the last dose. Men must avoid fathering a child during treatment with Alunbrig and for at least 3 months after taking the last dose.
If you are going to have surgery, tell the surgeon that you are taking this medicine.
If you or your partner becomes pregnant while taking Alunbrig, tell your doctor immediately.
Keep all of your doctor's appointments so that your progress can be checked. Your doctor will do blood tests and other tests from time to time to monitor your progress and check for side effects. If necessary, your doctor may decide to reduce your dose, temporarily interrupt your treatment or stop it altogether.
Things you must not do
Do not use this medicine to treat any other complaints unless your doctor tells you to.
Do not give this medicine to anyone else, even if they have the same condition as you.
Do not stop taking Alunbrig, or lower the dosage, without checking with your doctor first.
Things to be careful of
Be careful driving or operating machinery until you know how Alunbrig affects you. Alunbrig may cause visual disturbances, dizziness or tiredness. Do not drive or use machines during treatment if such signs occur.
Avoid any grapefruit products during treatment as they may change the amount of Alunbrig in your body.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Alunbrig.
It helps most people with non-small cell lung cancer, but it may have unwanted side effects in a few people. All medicines have some unwanted side effects. Sometimes they are serious, but most of the time they are not. You may need medical attention if you get some of the side-effects.
Taking Alunbrig may cause serious lung related side-effects in some people, particularly within the first week.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- lung or breathing problems including shortness of breath or difficulty breathing, cough and/or fever, especially within the first week of taking Alunbrig
- slow heart beat (bradycardia) – symptoms can include chest pain, dizziness, light headedness or fainting – or palpitations
- new or worsening signs and symptoms of muscle problems, including unexplained muscle pain that does not go away, tenderness or weakness
The above list includes very serious side effects. You may need urgent medical attention or hospitalisation.
Tell your doctor as soon as possible if you notice any of the following:
- tiredness, headaches, being short of breath when exercising, dizziness and looking pale
- passing large amounts of urine, excessive thirst and having a dry mouth
- difficulty sleeping
- numbness or weakness of the arms and legs
- change in sense of taste
- problems with your eyes such as blurred or impaired vision, black dots or white spots in your vision, double vision
- high blood pressure
- abdominal pain, nausea and/or vomiting
- diarrhoea or constipation
- rash and/or itching
- pain and swelling of the joints
- swelling throughout the body
- fever
- decrease in appetite or weight loss
The above list includes the more common side effects of your medicine, some of which may require medical attention.
Tell your doctor or pharmacist if you notice anything else that is making you feel unwell. Other side effects not listed above may occur in some people.
Some of these side effects can only be found when your doctor does tests. These tests could show a change in the liver, kidneys, levels of blood cells or changes in heart rate or blood pressure.
After using Alunbrig
Storage
Keep your tablets in the pack until it is time to take them. If you take the tablets out of the package they may not keep well.
Keep the medicine in a cool, dry place where the temperature stays below 30°C.
Do not store it or any other medicine in the bathroom, near a sink, or on a windowsill. Do not leave it in the car. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor or pharmacist tells you to stop taking this medicine, or the medicine has passed its expiry date, ask your pharmacist what to do with any that are left over.
Product description
What it looks like
Alunbrig 30 mg – white to off-white, round tablet, marked with a "U3" on one side.
Alunbrig 90 mg – white to off-white, oval tablet, marked with a "U7" on one side.
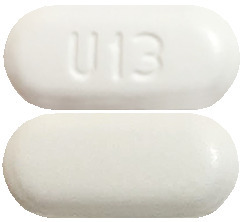
Alunbrig 180 mg – white to off-white, oval tablet, marked with a "U13" on one side.
The 30 mg, 90 mg and 180 mg strengths of Alunbrig are available in boxes of 28 tablets.
The one-month initiation pack contains 1 pack of 7 x 90 mg tablets and 21 x 180 mg tablets.
Ingredients
Alunbrig contains 30, 90 or 180 mg of brigatinib as the active ingredient:
It also contains
- lactose monohydrate
- microcrystalline cellulose
- sodium starch glycollate type A
- hydrophobic colloidal silica anhydrous
- magnesium stearate
- OPADRY II White (PI 11376)
Supplier
Alunbrig is supplied in Australia by:
Takeda Pharmaceuticals
Australia Pty Ltd
Level 52 Chifley Square
Sydney NSW 2000
Ph: 1800 675 957
® Registered Trademark
This leaflet was prepared in March 2019
Australian Registration Number(s)
30 mg tablets: AUST R 299965
90 mg tablets: AUST R 299968
180 mg tablets: AUST R 299964
One-month initiation pack: AUST R 299966
Version No. 1.0
Published by MIMS March 2020
 Permanently discontinue Alunbrig if patient is unable to tolerate the 60 mg once daily dose.
Permanently discontinue Alunbrig if patient is unable to tolerate the 60 mg once daily dose.

 The PFS for patients with CNS metastases at baseline (HR = 0.25, 95% CI: 0.14 0.46, median PFS for Alunbrig = 24 months, 95% CI: 18.37 NE, median PFS for crizotinib = 5.6 months, 95% CI: 3.84 9.4) and without CNS metastases at baseline (HR = 0.65, 95% CI: 0.44 0.97, median PFS for Alunbrig = 24 months, 95% CI: 15.67 NE, median PFS for crizotinib = 13 months, 95% CI: 9.46-21.13), indicated benefit of Alunbrig over crizotinib in both subgroups.
The PFS for patients with CNS metastases at baseline (HR = 0.25, 95% CI: 0.14 0.46, median PFS for Alunbrig = 24 months, 95% CI: 18.37 NE, median PFS for crizotinib = 5.6 months, 95% CI: 3.84 9.4) and without CNS metastases at baseline (HR = 0.65, 95% CI: 0.44 0.97, median PFS for Alunbrig = 24 months, 95% CI: 15.67 NE, median PFS for crizotinib = 13 months, 95% CI: 9.46-21.13), indicated benefit of Alunbrig over crizotinib in both subgroups. At the data cut off point overall survival data was not mature.
At the data cut off point overall survival data was not mature.



 In ALTA, 201 patients had at least 1 evaluable post-baseline assessment out of the 222 patients. Waterfall plots displaying the maximum decrease from baseline in the sum of the longest tumour diameters shows that the majority of patients treated with Alunbrig had a reduction in tumour burden in both the 90 mg and 180 mg regimens in ALTA (Figure 5 and Figure 6).
In ALTA, 201 patients had at least 1 evaluable post-baseline assessment out of the 222 patients. Waterfall plots displaying the maximum decrease from baseline in the sum of the longest tumour diameters shows that the majority of patients treated with Alunbrig had a reduction in tumour burden in both the 90 mg and 180 mg regimens in ALTA (Figure 5 and Figure 6).
 Of the 222 enrolled patients, baseline tumour tissue samples were evaluable in 17 patients. Responses were seen in patients with and without secondary ALK kinase domain mutations, including one patient with a secondary ALK kinase domain mutation of G1202R.
Of the 222 enrolled patients, baseline tumour tissue samples were evaluable in 17 patients. Responses were seen in patients with and without secondary ALK kinase domain mutations, including one patient with a secondary ALK kinase domain mutation of G1202R. In ALTA, patients overall experienced positive changes relative to baseline in quality-of-life (QOL) during treatment with brigatinib. The mean QOL, measured by the summary Global Health Status/QOL score of the European Organisation for Research and Treatment of Cancer (EORTC) Quality-of-Life Questionnaire (QLQ)-C30, was maintained above baseline mean values throughout follow-up (median: 17.9 months) across both dose groups.
In ALTA, patients overall experienced positive changes relative to baseline in quality-of-life (QOL) during treatment with brigatinib. The mean QOL, measured by the summary Global Health Status/QOL score of the European Organisation for Research and Treatment of Cancer (EORTC) Quality-of-Life Questionnaire (QLQ)-C30, was maintained above baseline mean values throughout follow-up (median: 17.9 months) across both dose groups. Brigatinib is an off-white to beige/tan solid. It is very slightly soluble in water, highly soluble from pH 1.5-6.5, slightly soluble in ethanol and soluble in methanol. The pKas were determined to be: 1.73 ± 0.02 (base), 3.65 ± 0.01 (base), 4.72 ± 0.01 (base), and 8.04 ± 0.01 (base).
Brigatinib is an off-white to beige/tan solid. It is very slightly soluble in water, highly soluble from pH 1.5-6.5, slightly soluble in ethanol and soluble in methanol. The pKas were determined to be: 1.73 ± 0.02 (base), 3.65 ± 0.01 (base), 4.72 ± 0.01 (base), and 8.04 ± 0.01 (base).