1. Why am I using AMGEVITA?
AMGEVITA contains the active ingredient adalimumab. AMGEVITA is a biosimilar medicine. It has been assessed to be as safe and effective as the reference product and provides the same health outcomes.
AMGEVITA is used to treat any of the following inflammatory conditions (listed in alphabetical order):
- Ankylosing spondylitis
- Crohn’s disease in adults and children aged 6 years and over
- Enthesitis-related arthritis
- Hidradenitis suppurativa in adults and adolescents aged 12 years and over
- Polyarticular juvenile idiopathic arthritis
- Psoriasis in adults and children aged 4 years and over
- Psoriatic arthritis
- Rheumatoid arthritis
- Ulcerative colitis, and
- Uveitis.
2. What should I know before I use AMGEVITA?
Warnings
Do not use AMGEVITA if any of the following apply:
- you are allergic to adalimumab, or any of the ingredients listed at the end of this leaflet. Always check the ingredients to make sure you can use it.
- you have a severe infection such as sepsis (a serious infection of the blood), tuberculosis (a serious infection of the lungs caused by bacteria), or other severe infection, caused by a virus, fungus, parasite or bacteria.
- you have heart failure considered by your doctor to be moderate or severe.
Check with your doctor if you have:
- any vaccinations scheduled
- any surgery planned
- a lung disease called chronic obstructive pulmonary disease (COPD)
- a disease that affects the insulating layer of the nerves, e.g. multiple sclerosis (MS)
- current active hepatitis B, have ever had hepatitis B, are a carrier of the hepatitis B virus or you think you may be at risk of contracting hepatitis B
- ever had tuberculosis, or you have been in close contact with someone who has tuberculosis. Tuberculosis can develop during therapy even if you have received treatment for the prevention of tuberculosis.
Check with your doctor if you have or have had:
- an infection that does not go away or keeps coming back, this can include leg ulcers
- an infection caused by a fungus, or you have lived or travelled in countries where fungal infections are common
- uveitis, where the middle layer of the eyeball is inflamed
- allergic reactions such as chest tightness, wheezing, dizziness, swelling or rash
- a blood disorder
- low resistance to disease
- a heart condition
- cancer or autoimmune disease
- kidney problems
- liver problems
- psoriasis (a skin disease that produces patches of thickened, scaly skin that is not contagious)
- phototherapy, also known as light therapy, for psoriasis.
Check with your doctor if you take any medicines for any other condition.
During treatment, you may be at risk of developing certain side effects. It is important you understand these risks and how to monitor for them. See additional information under Section 6. Are there any side effects?
Pregnancy
Make sure your doctor knows if you are pregnant or intend to become pregnant. AMGEVITA should only be used in pregnancy if clearly needed.
If you use AMGEVITA during pregnancy your baby may have a higher risk of getting an infection.
You should consider the use of effective contraception to prevent pregnancy and continue its use for at least 5 months after the last AMGEVITA injection.
Tell your baby’s doctors if you have taken AMGEVITA while you are pregnant, especially before your baby receives any vaccinations.
Breastfeeding
Make sure your doctor knows if you are breastfeeding or intend to breastfeed.
Use in children
Wherever possible, it is recommended that children are up to date with all vaccinations, according to current immunisation guidelines, before they are started on AMGEVITA treatment.
Treatment of Crohn’s disease in children should be supported by good nutrition to allow appropriate growth.
The long-term effects of AMGEVITA on the growth and development of children is not known.
Use in the elderly
If you are over 65, you may be more likely to get an infection while taking AMGEVITA.
3. What if I am taking other medicines?
Tell your doctor or pharmacist if you are taking any other:
- medicines
- vitamins or supplements that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interfere with AMGEVITA and affect how it works.
Do not take AMGEVITA if you are taking the following medicine:
- anakinra, a medicine used to treat rheumatoid arthritis, juvenile idiopathic arthritis and conditions associated with a defect in a protein called cryopyrin.
Medicines that may increase the risk of infections when taken with AMGEVITA include:
- anakinra
- abatacept, a medicine used to treat rheumatoid arthritis, polyarticular juvenile idiopathic arthritis, psoriatic arthritis
- azathioprine, a medicine used for suppressing the immune system to treat various conditions
- 6-mercaptopurine, a medicine used to treat certain types of leukaemia (a blood disorder).
AMGEVITA may affect how other medicines that you take work.
Check with your doctor or pharmacist if you are not sure about what medicines, vitamins or supplements you are taking and if these affect AMGEVITA.
4. How do I use AMGEVITA?
How much to use
Doses for each condition are given in the following text (in alphabetic order).
*For 160 mg and 80 mg doses, please also see Special dosing instructions (at the end of Section 4).
Ankylosing spondylitis in adults
- Inject one 40 mg dose every fortnight.
Crohn’s disease in adults
- Inject 160 mg* on day 1, followed by 80 mg* on day 15 and 40 mg on day 29.
- Then, continue to inject 40 mg every fortnight (maintenance dose). Your doctor may change the maintenance dose to 40 mg every week, or 80 mg* every fortnight, depending on your response.
Crohn’s disease in children
If the patient’s body weight is at least 40 kg:
- Inject 160 mg* on day 1, followed by 80 mg* on day 15 and 40 mg on day 29.
- Then, continue to inject 40 mg every fortnight (maintenance dose). Your doctor may change this maintenance dose to 40 mg every week, or 80 mg* every fortnight, depending on your response.
If the patient’s body weight is less than 40 kg:
- Inject 80 mg* on day 1, followed by 40 mg on day 15, and 20 mg on day 29.
- Then, continue to inject 20 mg every fortnight. Your doctor may change this maintenance dose to 20 mg every week, depending on your response.
Enthesitis-related arthritis
If the patient’s body weight is at least 30 kg:
- Inject one 40 mg dose every fortnight.
If the patient’s body weight is between 10 and 30 kg:
- Inject one 20 mg dose every fortnight.
Hidradenitis suppurativa in adults
- Inject 160 mg* on day 1, followed by 80 mg* on day 15.
- Then continue to inject 40 mg every week or 80 mg* every fortnight from day 29 (maintenance dose).
Hidradenitis suppurativa (HS) in adolescents
- Inject 80 mg* on day 1, followed by 40 mg on day 8, and 40 mg on day 22.
- Then continue to inject 40 mg every fortnight (maintenance dose). Your doctor may change this maintenance dose to 40 mg every week, or 80 mg* every fortnight depending on your response.
Use an antiseptic face wash on the affected areas.
Polyarticular juvenile idiopathic arthritis
If the patient’s body weight is at least 30 kg:
- Inject one 40 mg dose every fortnight.
If the patient’s body weight is between 10 and 30 kg:
- Inject one 20 mg dose every fortnight.
Psoriasis in adults
- Inject 80 mg* on day 1, followed by 40 mg on day 8 and 40 mg on day 22.
- Then, continue to inject 40 mg every fortnight (maintenance dose). Your doctor may change this maintenance dose to 40 mg every week, or 80 mg* every fortnight, depending on your response.
Psoriasis in children
If the patient’s body weight is at least 40 kg:
- Inject 40 mg on day 1, followed by 40 mg on day 8 and 40 mg on day 22.
- Then continue to inject 40 mg every fortnight (maintenance dose).
If the patient’s body weight is less than 40 kg:
- Inject 20 mg on day 1, inject 20 mg on day 8, then 20 mg on day 22.
- Then continue to inject 20 mg every fortnight (maintenance dose).
Psoriatic arthritis in adults
- Inject one 40 mg dose every fortnight.
Rheumatoid arthritis in adults
- Inject one 40 mg dose every fortnight.
If you are not taking methotrexate, your doctor may change this dose to 40 mg every week, or 80 mg* every fortnight, depending on your response.
Ulcerative colitis in adults
- Inject 160 mg* on day 1, followed by 80 mg* on day 15 and 40 mg on day 29.
- Then, continue to inject 40 mg every fortnight (maintenance dose). Your doctor may change this maintenance dose to 40 mg every week, or 80 mg* every fortnight, depending on your response.
Uveitis in adults
- Inject 80 mg* on day 1, followed by 40 mg on day 8 and 40 mg on day 22. Then continue to inject 40 mg every fortnight (maintenance dose).
Special dosing instructions
80 mg dose can be given as two 40 mg injections in 1 day.
160 mg dose can be given as either of the following:
- four 40 mg injections in 1 day, or
- two 40 mg injections per day over 2 consecutive days.
Some patients may need to use AMGEVITA and take other medicines. Your doctor will tell you which medicines to take, how to take them, and how long to take them.
How to use AMGEVITA
Read the Instructions for Use that are supplied in the pack, with the product, before preparing and using an AMGEVITA injection.
AMGEVITA is injected under the skin (subcutaneous). An injection should not be attempted until proper training has been received on the correct injection technique.
AMGEVITA can be injected by the patient, or by someone else, such as a family member, friend or carer.
Do not mix the solution for injection with any other medicine.
If you forget to use AMGEVITA
It is important that you use your medicine as prescribed by your doctor.
If you miss your dose at the usual time, inject AMGEVITA as soon as you remember, and continue injecting the next dose at the usual time on your scheduled day.
Do not take a double dose to make up for any dose you missed.
If you inject too much AMGEVITA
If you think that you have used/been given too much AMGEVITA, you may need urgent medical attention.
You should immediately:
- phone the Poisons Information Centre (by calling 13 11 26), or
- contact your doctor, or
- go to the Emergency Department at your nearest hospital.
You should do this even if there are no signs of discomfort or poisoning.
Always take the outer carton of the medicine with you.
5. What should I know while using AMGEVITA?
Things you should do
- AMGEVITA should be used regularly, as prescribed by your doctor.
- Follow all instructions given to you and use AMGEVITA until your doctor tells you to stop.
- Keep all your doctor’s appointments so your progress can be tracked.
- Keep your appointments for blood tests. Some side effects are seen in blood results before you have any symptoms.
- Check with your doctor before you receive any vaccines.
- Remind any healthcare professional you visit that you are using AMGEVITA, especially if you are scheduled for surgery or to receive any live vaccines (e.g. Bacille Calmette-Guerin or oral polio vaccine).
Call your doctor straight away if you:
- Get symptoms of an infection, such as a fever, skin sores, feeling tired, any problems with your teeth or gums or pain when passing urine or blood in your urine.
- Become pregnant while using AMGEVITA.
- Notice new skin lesions (skin spots or sores), or if existing lesions change appearance.
Things you should not do
- Do not stop using this medicine or change the dose without checking with your doctor.
Driving or using machines
Be careful before you drive or use any machines or tools until you know how AMGEVITA affects you.
Drinking alcohol
Tell your doctor if you drink alcohol. There is no information on the use of alcohol with AMGEVITA.
Looking after your medicine
Follow the instructions in the AMGEVITA carton for instructions on how to take care of your medicine properly.
Keep AMGEVITA in a refrigerator (at 2°C to 8°C). Do not freeze. Keep it in the carton protected from light.
Keep it where children cannot reach it.
When necessary, AMGEVITA may be stored at room temperature (25°C) for a maximum of 14 days, protected from light.
Once removed from the refrigerator, each pen or syringe must be used within 14 days or discarded, even if it has been returned to the refrigerator.
When to discard your medicine (as relevant)
After injecting AMGEVITA, immediately throw away the used syringe or pen into a special sharps container.
Discard any AMGEVITA that has been removed from the refrigerator for more than 14 days.
Getting rid of any unwanted medicine
If your doctor advises that you no longer need to use this medicine or it is out of date, take it to any pharmacy for safe disposal.
6. Are there any side effects?
All medicines can have side effects. If you do experience any side effects, most of them are minor and temporary. However, some side effects may need medical attention.
See the information below and, if you need to, ask your doctor or pharmacist if you have any further questions about side effects.
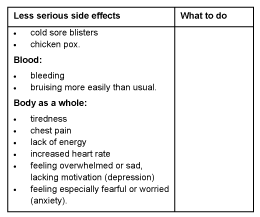
Less serious side effects

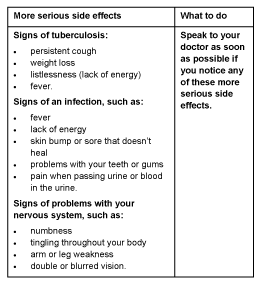
More serious side effects
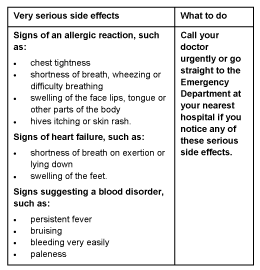
Very serious side effects
Tell your doctor or pharmacist if you notice anything else that may be making you feel unwell. Other side effects not listed here may occur in some people.
Reporting side effects
After you have received medical advice for any side effects you experience, you can report side effects to the Therapeutic Goods Administration online at www.tga.gov.au/ reporting-problems. By reporting side effects, you can help provide more information on the safety of this medicine.
Always make sure you speak to your doctor or pharmacist before you decide to stop taking any of your medicines.
7. Product details
This medicine is only available with a doctor's prescription.
What AMGEVITA contains
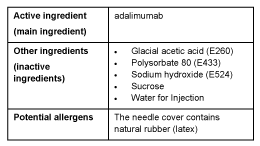
Do not take this medicine if you are allergic to any of these ingredients.
What AMGEVITA looks like
AMGEVITA is a clear, colourless, sterile solution containing:
- adalimumab 20 mg in 0.4 mL solution in a pre-filled syringe AUST R 278702)
- adalimumab 40 mg in 0.8 mL solution in a pre-filled syringe (AUST R 278701)
- adalimumab 40 mg in 0.8 mL solution in a pre-filled SureClick® pen (AUST R 273536).
Pre-filled syringes and pens are available in pack containing:
- 1 syringe, or
- 2 syringes, or
- 2 SureClick® pens.
Who distributes AMGEVITA
Amgen Australia Pty Ltd
ABN 31 051 057 428
Level 7, 123 Epping Road
North Ryde NSW 2113
Medical Information: 1800 803 638
[email protected]
www.amgen.com.au.
AMGEVITA® and SureClick® are registered trademarks of Amgen.
Version information
This leaflet was prepared in January 2021.
Version 1
Published by MIMS July 2021
 Available data suggest that a clinical response is usually achieved within 12 weeks of treatment. Continued therapy should be carefully reconsidered in a patient not responding within this time period.
Available data suggest that a clinical response is usually achieved within 12 weeks of treatment. Continued therapy should be carefully reconsidered in a patient not responding within this time period. Continued therapy should be carefully considered in a subject not responding by week 12.
Continued therapy should be carefully considered in a subject not responding by week 12. Some patients who experience a decrease in their response may benefit from an increase in dosage to 40 mg Amgevita every week or 80 mg fortnightly.
Some patients who experience a decrease in their response may benefit from an increase in dosage to 40 mg Amgevita every week or 80 mg fortnightly. During maintenance treatment, corticosteroids may be tapered in accordance with clinical practice guidelines. Some patients who experience decrease in their response may benefit from an increase in dosing frequency to 40 mg Amgevita every week, or 80 mg fortnightly.
During maintenance treatment, corticosteroids may be tapered in accordance with clinical practice guidelines. Some patients who experience decrease in their response may benefit from an increase in dosing frequency to 40 mg Amgevita every week, or 80 mg fortnightly. If retreatment with Amgevita is indicated, the above guidance on dose and treatment duration should be followed.
If retreatment with Amgevita is indicated, the above guidance on dose and treatment duration should be followed. In adolescent patients with inadequate response to adalimumab 40 mg fortnightly, an increase in dosage frequency to 40 mg every week or 80 mg fortnightly may be considered.
In adolescent patients with inadequate response to adalimumab 40 mg fortnightly, an increase in dosage frequency to 40 mg every week or 80 mg fortnightly may be considered. Antibiotics may be continued during treatment with adalimumab if necessary. Should treatment need to be interrupted, adalimumab may be re-introduced. In patients without any benefit after 12 weeks of treatment, therapy should be discontinued.
Antibiotics may be continued during treatment with adalimumab if necessary. Should treatment need to be interrupted, adalimumab may be re-introduced. In patients without any benefit after 12 weeks of treatment, therapy should be discontinued.


 The data in Table 12 reflects exposure to Amgevita/Amgevita in 152 subjects, Humira/Humira in 79 subjects, and Humira/Amgevita in 77 subjects in the Ps study treated at the recommended dose and schedule for a median of 1040 mg doses (see Section 5.1 Pharmacodynamic Properties, Clinical trials). 82.1% of subjects, from baseline to end of study, had at least 1 treatment emergent adverse event and similar proportions were reported across treatment groups (86.2% of subjects in Treatment Group A (Amgevita/Amgevita), 78.5% of subjects in Treatment Group B1 (Humira/Humira), and 85.7% of subjects in Treatment Group B2 (Humira/Amgevita).
The data in Table 12 reflects exposure to Amgevita/Amgevita in 152 subjects, Humira/Humira in 79 subjects, and Humira/Amgevita in 77 subjects in the Ps study treated at the recommended dose and schedule for a median of 1040 mg doses (see Section 5.1 Pharmacodynamic Properties, Clinical trials). 82.1% of subjects, from baseline to end of study, had at least 1 treatment emergent adverse event and similar proportions were reported across treatment groups (86.2% of subjects in Treatment Group A (Amgevita/Amgevita), 78.5% of subjects in Treatment Group B1 (Humira/Humira), and 85.7% of subjects in Treatment Group B2 (Humira/Amgevita).
 EC50 estimates ranging from 0.8 to 1.4 microgram/mL (microgram/mL) were obtained through PK/PD modelling of swollen joint count, tender joint count and ACR20 response from patients participating in Phase II and III trials.
EC50 estimates ranging from 0.8 to 1.4 microgram/mL (microgram/mL) were obtained through PK/PD modelling of swollen joint count, tender joint count and ACR20 response from patients participating in Phase II and III trials. Patients receiving adalimumab 40 mg every week in RA study II also achieved statistically significant ACR20, 50 and 70 response rates of 53.4%, 35.0% and 18.4%, respectively, at six months (see Figure 2).
Patients receiving adalimumab 40 mg every week in RA study II also achieved statistically significant ACR20, 50 and 70 response rates of 53.4%, 35.0% and 18.4%, respectively, at six months (see Figure 2). The results of the components of the ACR response criteria for RA study III are shown in Figure 3 and Table 14. ACR response rates and improvement in all ACR response criteria were maintained to week 104. Over the 2 years in RA study III, 20% of adalimumab patients achieved a major clinical response, defined as maintenance of an ACR70 response over a > 6 month period.
The results of the components of the ACR response criteria for RA study III are shown in Figure 3 and Table 14. ACR response rates and improvement in all ACR response criteria were maintained to week 104. Over the 2 years in RA study III, 20% of adalimumab patients achieved a major clinical response, defined as maintenance of an ACR70 response over a > 6 month period. In RA study III, 84.7% of patients with ACR20 responses at week 24 maintained the response at 52 weeks. Clinical responses were maintained for up to 5 years in the open-label portion of RA study III. ACR responses observed at week 52 were maintained or increased through 5 years of continuous treatment with 22% (115/534) of patients achieving major clinical response. A total of 372 (67.8%) subjects had no change in their MTX dose during the study, 141 (25.7%) subjects had a dose reduction and 36 (6.6%) subjects required a dose increase. A total of 149 (55.6%) subjects had no change in their corticosteroid dose during the study, 80 (29.9%) subjects had a dose reduction and 39 (14.6%) subjects required a dose increase. Figure 2 and Figure 3 illustrate the durability of ACR20 responses to adalimumab in RA Studies II and III.
In RA study III, 84.7% of patients with ACR20 responses at week 24 maintained the response at 52 weeks. Clinical responses were maintained for up to 5 years in the open-label portion of RA study III. ACR responses observed at week 52 were maintained or increased through 5 years of continuous treatment with 22% (115/534) of patients achieving major clinical response. A total of 372 (67.8%) subjects had no change in their MTX dose during the study, 141 (25.7%) subjects had a dose reduction and 36 (6.6%) subjects required a dose increase. A total of 149 (55.6%) subjects had no change in their corticosteroid dose during the study, 80 (29.9%) subjects had a dose reduction and 39 (14.6%) subjects required a dose increase. Figure 2 and Figure 3 illustrate the durability of ACR20 responses to adalimumab in RA Studies II and III.
 In the open-label extension for RA study V, ACR responses were maintained when followed for up to 10 years. However, no statistical hypothesis was tested in the OLE period. Of 542 patients who were randomised to adalimumab 40 mg fortnightly, 170 patients continued on adalimumab 40 mg fortnightly for 10 years. Among those, 154 patients (90.6%) had ACR20 responses; 127 patients (74.7%) had ACR50 responses and 102 patients (60.0%) had ACR70 responses.
In the open-label extension for RA study V, ACR responses were maintained when followed for up to 10 years. However, no statistical hypothesis was tested in the OLE period. Of 542 patients who were randomised to adalimumab 40 mg fortnightly, 170 patients continued on adalimumab 40 mg fortnightly for 10 years. Among those, 154 patients (90.6%) had ACR20 responses; 127 patients (74.7%) had ACR50 responses and 102 patients (60.0%) had ACR70 responses.
 In RA study V, adalimumab-treated patients had a mean duration of RA of less than 9 months and had not previously received MTX. Structural joint damage was assessed radiographically and expressed as change in modified total sharp score. The week 52 results are shown in Table 18. A statistically significant difference for change in modified Total Sharp Score and the erosion score was observed at week 52 and maintained at week 104.
In RA study V, adalimumab-treated patients had a mean duration of RA of less than 9 months and had not previously received MTX. Structural joint damage was assessed radiographically and expressed as change in modified total sharp score. The week 52 results are shown in Table 18. A statistically significant difference for change in modified Total Sharp Score and the erosion score was observed at week 52 and maintained at week 104. In the open-label extension of RA study V, the mean change from baseline at Year 10 in the modified Total Sharp Score was 10.8, 9.2 and 3.9 in patients originally randomised to methotrexate monotherapy, adalimumab monotherapy and adalimumab/methotrexate combination therapy, respectively. The corresponding proportions of patients with no radiographic progression were 31.3%, 23.7% and 36.7%, respectively.
In the open-label extension of RA study V, the mean change from baseline at Year 10 in the modified Total Sharp Score was 10.8, 9.2 and 3.9 in patients originally randomised to methotrexate monotherapy, adalimumab monotherapy and adalimumab/methotrexate combination therapy, respectively. The corresponding proportions of patients with no radiographic progression were 31.3%, 23.7% and 36.7%, respectively. At Year 2, 45.4% (94/207) of patients who originally entered the study achieved a -0.5 reduction in HAQ. 79.5% (115/195) of the patients who achieved a reduction in HAQ of -0.5 at the end of one year of adalimumab treatment maintained this response over 5 years of active treatment.
At Year 2, 45.4% (94/207) of patients who originally entered the study achieved a -0.5 reduction in HAQ. 79.5% (115/195) of the patients who achieved a reduction in HAQ of -0.5 at the end of one year of adalimumab treatment maintained this response over 5 years of active treatment.

 In subjects treated with adalimumab with no radiographic progression from baseline to week 48 (n = 102), 84% continued to show no radiographic progression through 144 weeks of treatment.
In subjects treated with adalimumab with no radiographic progression from baseline to week 48 (n = 102), 84% continued to show no radiographic progression through 144 weeks of treatment.
 A low level of disease activity [defined as a value < 20 (on a scale of 0-100 mm) in each of the four ASAS response parameters] was achieved at 24 weeks in 22% of adalimumab-treated patients vs. 6% in placebo-treated patients (p < 0.001). See Table 24.
A low level of disease activity [defined as a value < 20 (on a scale of 0-100 mm) in each of the four ASAS response parameters] was achieved at 24 weeks in 22% of adalimumab-treated patients vs. 6% in placebo-treated patients (p < 0.001). See Table 24. Results of this study were similar to those seen in the second randomised trial (AS study II or M03-606), a multicentre, double-blind, placebo-controlled study of 82 patients with AS. Patient Reported Outcomes (PROs) were assessed in both AS studies using the generic health status questionnaire SF-36 and the disease specific AS Quality of Life Questionnaire (ASQoL). The adalimumab-treated patients had significantly greater improvement in SF-36 Physical Component Score (mean change: 6.93) compared to placebo-treated patients (mean change: 1.55; p < 0.001) at week 12, which was maintained through week 24.
Results of this study were similar to those seen in the second randomised trial (AS study II or M03-606), a multicentre, double-blind, placebo-controlled study of 82 patients with AS. Patient Reported Outcomes (PROs) were assessed in both AS studies using the generic health status questionnaire SF-36 and the disease specific AS Quality of Life Questionnaire (ASQoL). The adalimumab-treated patients had significantly greater improvement in SF-36 Physical Component Score (mean change: 6.93) compared to placebo-treated patients (mean change: 1.55; p < 0.001) at week 12, which was maintained through week 24.
 Clinical remission results presented in Table 27 remained relatively constant irrespective of previous TNF-antagonist exposure. Of those in response at week 4 who attained remission during the study, patients in adalimumab maintenance groups maintained remission for a significantly longer time that patients in the placebo maintenance group (see Figure 5). Among patients who were not in response by week 12, therapy continued beyond 12 weeks did not result in significantly more responses. The group that received adalimumab every week did not show significantly higher remission rates than the group that received adalimumab fortnightly.
Clinical remission results presented in Table 27 remained relatively constant irrespective of previous TNF-antagonist exposure. Of those in response at week 4 who attained remission during the study, patients in adalimumab maintenance groups maintained remission for a significantly longer time that patients in the placebo maintenance group (see Figure 5). Among patients who were not in response by week 12, therapy continued beyond 12 weeks did not result in significantly more responses. The group that received adalimumab every week did not show significantly higher remission rates than the group that received adalimumab fortnightly. One hundred and seventeen (117) out of 854 patients had draining fistulas both at screening and at baseline. For the assessment of fistula healing, the data for both doses of adalimumab used in the study were pooled. The proportion of subjects (ITT population) with fistula healing at week 26 was statistically significantly greater in patients treated with adalimumab [21/70 (30.0%)] compared to placebo [6/47 (12.8%)]. Complete fistula healing was maintained through week 56 in 23/70 (32.9%) and 6/47 (12.8%) patients (ITT population) in the adalimumab and placebo groups, respectively.
One hundred and seventeen (117) out of 854 patients had draining fistulas both at screening and at baseline. For the assessment of fistula healing, the data for both doses of adalimumab used in the study were pooled. The proportion of subjects (ITT population) with fistula healing at week 26 was statistically significantly greater in patients treated with adalimumab [21/70 (30.0%)] compared to placebo [6/47 (12.8%)]. Complete fistula healing was maintained through week 56 in 23/70 (32.9%) and 6/47 (12.8%) patients (ITT population) in the adalimumab and placebo groups, respectively.

 Rates of discontinuation of corticosteroids or immunomodulators and fistula remission (defined as a closure of all fistulas that were draining at baseline for at least 2 consecutive post-baseline visits) are presented in Table 30.
Rates of discontinuation of corticosteroids or immunomodulators and fistula remission (defined as a closure of all fistulas that were draining at baseline for at least 2 consecutive post-baseline visits) are presented in Table 30. Statistically significant increases (improvement) from baseline to week 26 and 52 in Body Mass Index and height velocity were observed for both treatment groups. Statistically and clinically significant improvements from baseline were also observed in both treatment groups for quality of life parameters (including IMPACT III).
Statistically significant increases (improvement) from baseline to week 26 and 52 in Body Mass Index and height velocity were observed for both treatment groups. Statistically and clinically significant improvements from baseline were also observed in both treatment groups for quality of life parameters (including IMPACT III). Adalimumab should be discontinued in patients who do not achieve a clinical response during the first 8 weeks of therapy because very few patients will achieve clinical remission with continuing treatment. In both UC studies, of patients given adalimumab 160/80 mg at baseline who did not achieve a clinical response at week 8, 5.2%, and 17.0% went on to be in remission and response, respectively at week 52. See Table 32.
Adalimumab should be discontinued in patients who do not achieve a clinical response during the first 8 weeks of therapy because very few patients will achieve clinical remission with continuing treatment. In both UC studies, of patients given adalimumab 160/80 mg at baseline who did not achieve a clinical response at week 8, 5.2%, and 17.0% went on to be in remission and response, respectively at week 52. See Table 32. Statistically significant reductions of both all-cause and UC-related rates of hospitalisation were observed in a pooled analysis of studies UC I and II.
Statistically significant reductions of both all-cause and UC-related rates of hospitalisation were observed in a pooled analysis of studies UC I and II.
 Two of the continuous treatment populations entering trial M03-658 were those from Period C of study I and those from study II.
Two of the continuous treatment populations entering trial M03-658 were those from Period C of study I and those from study II.
 Patients who achieved PASI 75 and PGA clear or minimal were withdrawn from treatment for up to 36 weeks and monitored for loss of disease control (loss of PGA response). Patients were then re-treated with adalimumab 0.8 mg/kg fortnightly for an additional 16 weeks. Among patients who were responders to the initial 16 weeks of treatment but who relapsed upon withdrawal and were retreated, PASI 75 response of 78.9% (15 of 19 subjects) and PGA clear or minimal of 52.6% (10 of 19 subjects) was observed.
Patients who achieved PASI 75 and PGA clear or minimal were withdrawn from treatment for up to 36 weeks and monitored for loss of disease control (loss of PGA response). Patients were then re-treated with adalimumab 0.8 mg/kg fortnightly for an additional 16 weeks. Among patients who were responders to the initial 16 weeks of treatment but who relapsed upon withdrawal and were retreated, PASI 75 response of 78.9% (15 of 19 subjects) and PGA clear or minimal of 52.6% (10 of 19 subjects) was observed.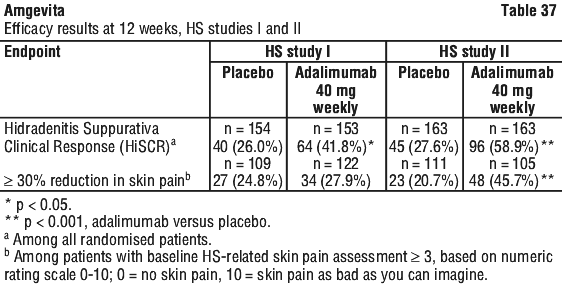 There is a statistically significantly higher HiSCR rate at week 36 in patients who continued to receive weekly adalimumab compared to those who stopped adalimumab at week 12.
There is a statistically significantly higher HiSCR rate at week 36 in patients who continued to receive weekly adalimumab compared to those who stopped adalimumab at week 12.


 Additionally, in study UV I, statistically significant differences in favour of adalimumab versus placebo were observed for the secondary endpoints changes in AC cell grade, vitreous haze grade, and logMAR BCVA (mean change from best state prior to week 6 to the final visit; p values: 0.011, < 0.001 and 0.003, respectively).
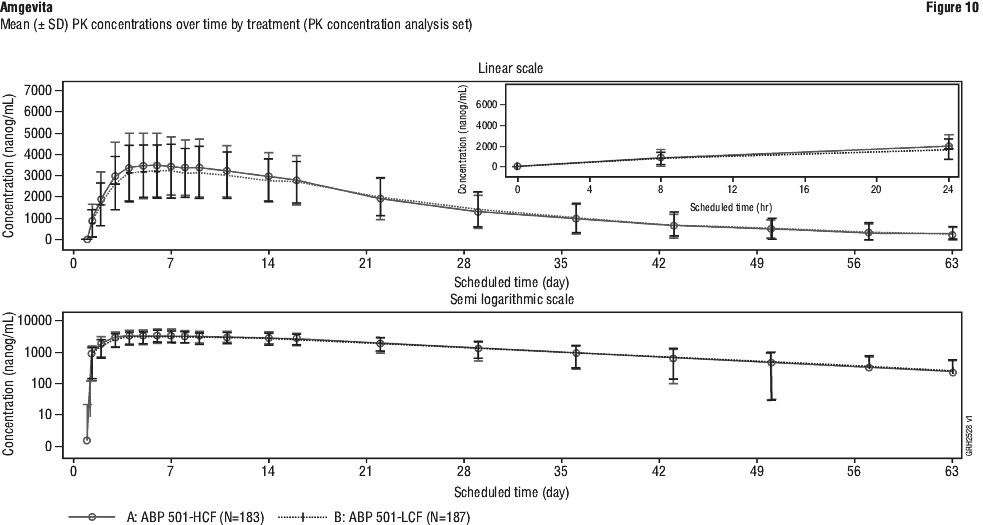
Additionally, in study UV I, statistically significant differences in favour of adalimumab versus placebo were observed for the secondary endpoints changes in AC cell grade, vitreous haze grade, and logMAR BCVA (mean change from best state prior to week 6 to the final visit; p values: 0.011, < 0.001 and 0.003, respectively). The RR of ACR 20 primary endpoint was within the pre-specified margin and showed clinical equivalence between Amgevita and Humira. The results of the components of the ACR response criteria for RA ABP-study 1 are shown in Table 41. ACR response rates and improvement in all components of ACR response showed an absence of clinically meaningful differences between the two groups at week 24. The time course of ACR20 response is shown in Figure 8.
The RR of ACR 20 primary endpoint was within the pre-specified margin and showed clinical equivalence between Amgevita and Humira. The results of the components of the ACR response criteria for RA ABP-study 1 are shown in Table 41. ACR response rates and improvement in all components of ACR response showed an absence of clinically meaningful differences between the two groups at week 24. The time course of ACR20 response is shown in Figure 8.

 The primary endpoint was PASI percent improvement from baseline to week 16. At week 16, the PASI percent improvement from baseline was 80.9 in the Amgevita group and 83.1 in the Humira group. The least-squares (LS) mean difference of PASI percent improvement from baseline to week 16 between Amgevita and Humira was -2.18 with the 2 sided 95% CI of (-7.39, 3.02). The 95% CI was within the predefined equivalence margin (-15, 15), thus demonstrating clinical equivalence of Amgevita and Humira.
The primary endpoint was PASI percent improvement from baseline to week 16. At week 16, the PASI percent improvement from baseline was 80.9 in the Amgevita group and 83.1 in the Humira group. The least-squares (LS) mean difference of PASI percent improvement from baseline to week 16 between Amgevita and Humira was -2.18 with the 2 sided 95% CI of (-7.39, 3.02). The 95% CI was within the predefined equivalence margin (-15, 15), thus demonstrating clinical equivalence of Amgevita and Humira.
 Following the administration of adalimumab 24 mg/m2 (up to a maximum of adalimumab 40 mg) subcutaneously fortnightly to patients with ERA, the mean trough steady-state (values measured at week 24) serum adalimumab concentrations were 8.8 ± 6.6 microgram/mL for adalimumab without concomitant MTX and 11.8 ± 4.3 microgram/mL with concomitant MTX. Based on a population pharmacokinetic (PK) modelling approach, simulated steady-state adalimumab serum trough concentrations for a weight-based dosing regimen (adalimumab 20 mg fortnightly for body weight < 30 kg and adalimumab 40 mg fortnightly for body weight ≥ 30 kg) were comparable to the simulated trough concentrations for the body surface area-based regimen.
Following the administration of adalimumab 24 mg/m2 (up to a maximum of adalimumab 40 mg) subcutaneously fortnightly to patients with ERA, the mean trough steady-state (values measured at week 24) serum adalimumab concentrations were 8.8 ± 6.6 microgram/mL for adalimumab without concomitant MTX and 11.8 ± 4.3 microgram/mL with concomitant MTX. Based on a population pharmacokinetic (PK) modelling approach, simulated steady-state adalimumab serum trough concentrations for a weight-based dosing regimen (adalimumab 20 mg fortnightly for body weight < 30 kg and adalimumab 40 mg fortnightly for body weight ≥ 30 kg) were comparable to the simulated trough concentrations for the body surface area-based regimen.