1 Name of Medicine
Vutrisiran.
2 Qualitative and Quantitative Composition
Each pre-filled syringe contains vutrisiran sodium equivalent to 25 mg vutrisiran in 0.5 mL solution.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Solution for injection.
Amvuttra is a clear, colourless to yellow sterile solution practically free from visible particles.
4.1 Therapeutic Indications
Amvuttra is indicated for the treatment of hereditary transthyretin-mediated amyloidosis (hATTR amyloidosis) in adult patients with stage 1 or stage 2 polyneuropathy.
4.2 Dose and Method of Administration
Therapy should be initiated under the supervision of a physician knowledgeable in the management of amyloidosis. Treatment should be started as early as possible in the disease course to prevent the accumulation of disability.
Dosage.
The recommended dose of Amvuttra is 25 mg administered via subcutaneous injection once every 3 months.
Vitamin A supplementation at approximately, but not exceeding, 2500 IU to 3000 IU vitamin A per day is advised for patients treated with Amvuttra (see Section 4.4 Special Warnings and Precautions for Use).
The decision to continue treatment in those patients whose disease progresses to stage 3 polyneuropathy should be taken at the discretion of the physician based on the overall benefit and risk assessment.
Missed dose.
If a dose is missed, Amvuttra should be administered as soon as possible. Dosing should be resumed every 3 months, from the most recently administered dose.
Dosage adjustment.
Elderly patients.
No dose adjustment is required in patients ≥ 65 years of age (see Section 5.2 Pharmacokinetic Properties).
Hepatic impairment.
No dose adjustment is necessary in patients with mild hepatic impairment (total bilirubin ≤ 1 x upper limit of normal (ULN) and aspartate aminotransferase (AST) > 1 x ULN, or total bilirubin > 1.0 to 1.5 x ULN and any AST).
Renal impairment.
No dose adjustment is necessary in patients with mild or moderate renal impairment (estimated glomerular filtration rate [eGFR] ≥ 30 to < 90 mL/min/1.73 m2).
Paediatric population.
The safety and efficacy of Amvuttra in children or adolescents < 18 years of age have not been established. No data are available.
Method of administration.
Amvuttra is a solution for subcutaneous injection only. Amvuttra should be administered by a healthcare professional.
This medicinal product is ready-to-use and for single-use only.
Visually inspect the solution for particulate matter and discolouration. Do not use if discoloured or if particles are present.
Prior to administration, if stored cold, the pre-filled syringe should be allowed to warm by leaving carton at room temperature for about 30 minutes.
The subcutaneous injection should be administered into one of the following sites: the abdomen, thighs, or upper arms. Amvuttra should not be injected into scar tissue or areas that are reddened, inflamed, or swollen.
If injecting into the abdomen, the area around the navel should be avoided.4.3 Contraindications
Severe hypersensitivity (e.g. anaphylaxis) to the active substance or to any of the excipients listed, see Section 6.1 List of Excipients.
4.4 Special Warnings and Precautions for Use
Vitamin A deficiency.
By reducing serum transthyretin (TTR) protein, Amvuttra treatment leads to a decrease in serum vitamin A (retinol) levels (see Section 5.1 Pharmacodynamic Properties). Serum vitamin A levels below the lower limit of normal should be corrected and any ocular symptoms or signs due to vitamin A deficiency should be evaluated prior to initiation of treatment with Amvuttra.
Patients receiving Amvuttra should take oral supplementation of approximately, but not exceeding, 2500 IU to 3000 IU vitamin A per day to reduce the potential risk of ocular symptoms due to vitamin A deficiency. Ophthalmological assessment is recommended if patients develop ocular symptoms suggestive of vitamin A deficiency, including reduced night vision or night blindness, persistent dry eyes, eye inflammation, corneal inflammation or ulceration, corneal thickening or corneal perforation.
During the first 60 days of pregnancy, both too high or too low vitamin A levels may be associated with an increased risk of fetal malformation. Therefore, pregnancy should be excluded before initiating Amvuttra and women of childbearing potential should practice effective contraception (see Section 4.6 Fertility, Pregnancy and Lactation). If a woman intends to become pregnant, Amvuttra and vitamin A supplementation should be discontinued and serum vitamin A levels should be monitored and have returned to normal before conception is attempted. Serum vitamin A levels may remain reduced for more than 12 months after the last dose of Amvuttra.
In the event of an unplanned pregnancy, Amvuttra should be discontinued (see Section 4.6 Fertility, Pregnancy and Lactation). No recommendation can be given whether to continue or discontinue vitamin A supplementation during the first trimester of an unplanned pregnancy. If vitamin A supplementation is continued, the daily dose should not exceed 3000 IU per day, due to the lack of data supporting higher doses. Thereafter, vitamin A supplementation of 2500 IU to 3000 IU per day should be resumed in the second and third trimesters if serum vitamin A levels have not yet returned to normal, because of the increased risk of vitamin A deficiency in the third trimester.
It is not known whether vitamin A supplementation in pregnancy will be sufficient to prevent vitamin A deficiency if the pregnant female continues to receive Amvuttra. However, increasing vitamin A supplementation to above 3000 IU per day during pregnancy is unlikely to correct plasma retinol levels due to the mechanism of action of Amvuttra and may be harmful to the mother and fetus.
Use in hepatic impairment.
Vutrisiran has not been studied in patients with moderate or severe hepatic impairment and should only be used in these patients if the anticipated clinical benefit outweighs the potential risk (see Section 5.2 Pharmacokinetic Properties).
Use in renal impairment.
Vutrisiran has not been studied in patients with severe renal impairment or end-stage renal disease and should only be used in these patients if the anticipated clinical benefit outweighs the potential risk (see Section 5.2 Pharmacokinetic Properties).
Use in the elderly.
There are no special precautions for the use of Amvuttra in the elderly.
Paediatric use.
No data are available.
Effects on laboratory tests.
Serum TTR is a carrier of retinol binding protein, which facilitates transport of vitamin A in the blood. Treatment with Amvuttra reduces serum TTR levels, which results in reduced levels of retinol binding protein and vitamin A in the serum. However, transport and tissue uptake of vitamin A can occur through alternative mechanisms in the absence of retinol binding protein. As a result, during treatment with Amvuttra, laboratory tests for serum vitamin A do not reflect the total amount of vitamin A in the body and should not be used to guide vitamin A supplementation (see Section 4.4 Special Warnings and Precautions for Use; Section 5.1 Pharmacodynamic Properties).
Within the clinical study HELIOS-A, blood alkaline phosphatase (ALP) levels in the vutrisiran-treated patients showed a mean percent change of 36.71% from baseline, with mean ALP values remaining within normal reference range. Aggregate analysis of clinical studies involving Amvuttra, and additional post-marketing monitoring has not yet determined a clinically meaningful effect.4.5 Interactions with Other Medicines and Other Forms of Interactions
No clinical interaction studies have been performed. Vutrisiran is not expected to cause interactions or to be affected by inhibitors or inducers of cytochrome P450 enzymes, or to modulate the activity of transporters. Therefore, vutrisiran is not expected to have clinically significant interactions with other medicinal products.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
There are no data on the effects of Amvuttra on human fertility. Subcutaneous administration of vutrisiran by weekly doses up to 70 mg/kg in male and female rats (up to 2025-times and 1136- times the plasma AUC at the maximum recommended human dose of 25 mg every 3 months, respectively) through Gestation Day 6 or weekly doses of a rodent-specific surrogate at 30 mg/kg in female rats prior to and during mating, and continuing in females during gestation once weekly on Gestation Days 6, 12, and 17, resulted in no adverse effects upon the male or female fertility endpoints evaluated.
(Category D)
Treatment with Amvuttra reduces serum levels of vitamin A. Both too high or too low vitamin A levels may be associated with an increased risk of fetal malformation. Therefore, pregnancy should be excluded before initiation of treatment and women of childbearing potential should use effective contraception. If a woman intends to become pregnant, Amvuttra and vitamin A supplementation should be discontinued and serum vitamin A levels should be monitored and have returned to normal before conception is attempted (see Section 4.4 Special Warnings and Precautions for Use). Serum vitamin A levels may remain reduced for more than 12 months after the last dose of treatment.
There are no data on the use of Amvuttra in pregnant women. Animal studies are insufficient with respect to reproductive and developmental toxicity as vutrisiran is not pharmacologically active in rats and rabbits. Due to the potential teratogenic risk arising from unbalanced vitamin A levels, Amvuttra should not be used during pregnancy. As a precautionary measure, vitamin A (see Section 4.4 Special Warnings and Precautions for Use) and thyroid stimulating hormone levels should be obtained early in pregnancy. Close monitoring of the fetus should be carried out, especially during the first trimester.
It is unknown whether vutrisiran is excreted in human milk. There is insufficient information on the excretion of vutrisiran in animal milk.
A decision must be made whether to discontinue breastfeeding or to discontinue/abstain from Amvuttra, taking into account the benefit of breastfeeding for the child and the benefit of therapy for the woman.4.7 Effects on Ability to Drive and Use Machines
Amvuttra has no or negligible influence on the ability to drive and use machines.
4.8 Adverse Effects (Undesirable Effects)
Summary of the safety profile.
During the 18-month treatment period of the global, randomised, open-label clinical study (HELIOS-A) in adult patients with hATTR amyloidosis with polyneuropathy, the most frequently occurring adverse reactions reported in Amvuttra-treated patients were pain in extremity (15%) and arthralgia (11%).
Tabulated list of adverse reactions.
The adverse reactions are presented as MedDRA preferred terms and under the MedDRA System Organ Class (SOC). The frequency of the adverse reactions is expressed according to the following categories: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1 000 to < 1/100). See Table 1.
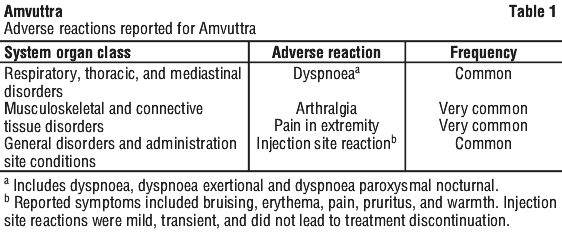
Description of selected adverse reactions.
Immunogenicity.
During the HELIOS-A 18-month treatment period, 4 (3.3%) Amvuttra-treated patients developed anti-drug antibodies (ADA). ADA titres were low and transient with no evidence of an effect on clinical efficacy, safety, or pharmacokinetic or pharmacodynamic profiles of vutrisiran.
Reporting suspected adverse reactions.
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
In case of overdose, it is recommended that the patient be monitored as medically indicated for any signs or symptoms of adverse reactions and appropriate symptomatic treatment be instituted.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Other Nervous System Drugs; ATC code: N07XX18.
Mechanism of action.
Amvuttra contains vutrisiran, a chemically stabilised double-stranded small interfering ribonucleic acid (siRNA) that specifically targets variant and wild-type transthyretin (TTR) messenger RNA (mRNA) and is covalently linked to a ligand containing three Nacetylgalactosamine (GalNAc) residues to enable delivery of the siRNA to hepatocytes.
Through a natural process called RNA interference (RNAi), vutrisiran causes the catalytic degradation of TTR mRNA in the liver, resulting in the reduction of variant and wild-type serum TTR protein levels.
Pharmacodynamic effects.
Mean serum TTR was reduced as early as Day 22, with mean near to steady state TTR reduction of 73% by Week 6. With repeat dosing of 25 mg once every 3 months, mean reductions of serum TTR after 9 and 18 months of treatment were 83% and 88%, respectively. Similar TTR reductions were observed regardless of genotype (V30M or non-V30M), prior TTR stabiliser use, weight, sex, age, or race.
Serum TTR is a carrier of retinol binding protein 4, which is the principal carrier of vitamin A in the blood. Amvuttra decreased vitamin A levels with mean steady state peak and trough reductions of 70% and 63%, respectively (see Section 4.4 Special Warnings and Precautions for Use; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Clinical trials.
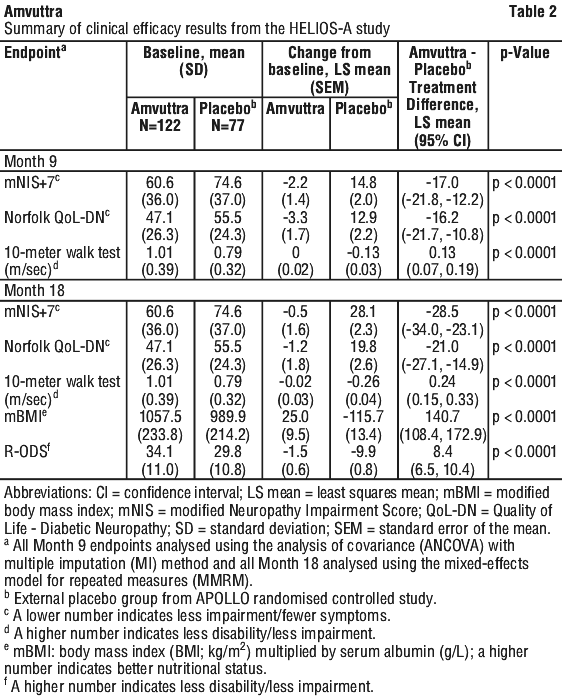
The efficacy of Amvuttra was studied in a global, randomised, open-label clinical study (HELIOS-A) in adult patients with hATTR amyloidosis with polyneuropathy. Patients were randomised 3:1 to receive 25 mg of Amvuttra (N=122) subcutaneously once every 3 months, or 0.3 mg/kg patisiran (N=42) intravenously once every 3 weeks. The treatment period of the study was conducted over 18 months with two analyses at Month 9 and at Month 18. Ninety-seven percent (97%) of Amvuttra-treated patients completed at least 18 months of the assigned treatments (vutrisiran or patisiran). Efficacy assessments were based on a comparison of the vutrisiran arm of the study with an external placebo group (placebo arm of the APOLLO Phase 3 study) comprised of a similar population of patients with hATTR amyloidosis with polyneuropathy. Assessment of non-inferiority of serum TTR reduction was based on comparison of the vutrisiran arm to the within-study patisiran arm.
Of the patients who received Amvuttra, the median patient age at baseline was 60 years (range 34 to 80 years), 38% were ≥ 65 years old, and 65% of patients were male. Twenty-two (22) different TTR variants were represented: V30M (44%), T60A (13%), E89Q (8%), A97S (6%), S50R (4%), V122I (3%), L58H (3%), and Other (18%). Twenty percent (20%) of patients had the V30M genotype and early onset of symptoms (< 50 years old). At baseline, 69% of patients had stage 1 disease (unimpaired ambulation; mild sensory, motor, and autonomic neuropathy in the lower limbs), and 31% had stage 2 disease (assistance with ambulation required; moderate impairment of the lower limbs, upper limbs, and trunk). There were no patients with stage 3 disease. Sixty-one percent (61%) of patients had prior treatment with TTR tetramer stabilisers. According to the New York Heart Association (NYHA) classification of heart failure, 9% of patients had class I and 35% had class II. Thirty-three percent (33%) of patients met pre-defined criteria for cardiac involvement (baseline LV wall thickness ≥ 13 mm with no history of hypertension or aortic valve disease).
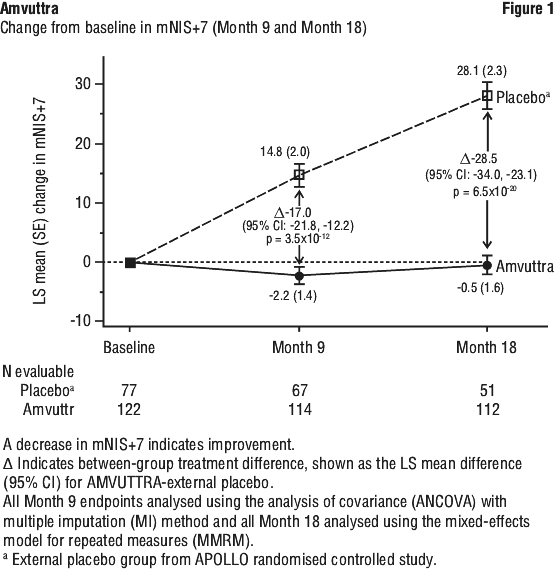
The primary efficacy endpoint was the change from baseline to Month 18 in modified Neuropathy Impairment Score +7 (mNIS+7). This endpoint is a composite measure of motor, sensory, and autonomic neuropathy including assessments of motor strength, reflexes, quantitative sensory testing, nerve conduction studies, and postural blood pressure, with the score ranging from 0 to 304 points, where an increasing score indicates worsening impairment.
The change from baseline to Month 18 in Norfolk Quality of Life-Diabetic Neuropathy (QoL-DN) total score was assessed as a secondary endpoint. The Norfolk QoL-DN questionnaire (patient-reported) includes domains relating to small fibre, large fibre, and autonomic nerve function, symptoms of polyneuropathy, and activities of daily living, with the total score ranging from -4 to 136, where increasing score indicates worsening quality of life.
Other secondary endpoints included gait speed (10-meter walk test), nutritional status (mBMI), and patient-reported ability to perform activities of daily living and social participation (Rasch-Built Overall Disability Scale [R-ODS]).
Treatment with Amvuttra in the HELIOS-A study demonstrated statistically significant improvements in all endpoints (see Table 2 and Figure 1) measured from baseline to Month 9 and 18, compared to the external placebo group of the APOLLO study (all p < 0.0001).
The time-averaged trough TTR percent reduction through Month 18 was 84.7% for vutrisiran and 80.6% for patisiran. The percent reduction in serum TTR levels in the vutrisiran arm was non-inferior (according to predefined criteria) to the within-study patisiran arm through Month 18 with a median difference of 5.3% (95% CI 1.2%, 9.3%).
 Patients receiving Amvuttra experienced similar benefit relative to placebo in mNIS+7 and Norfolk QoL-DN total score at Month 9 and Month 18 across all subgroups including age, sex, race, region, NIS score, V30M genotype status, prior TTR stabiliser use, disease stage, and patients with or without pre-defined criteria for cardiac involvement.
Patients receiving Amvuttra experienced similar benefit relative to placebo in mNIS+7 and Norfolk QoL-DN total score at Month 9 and Month 18 across all subgroups including age, sex, race, region, NIS score, V30M genotype status, prior TTR stabiliser use, disease stage, and patients with or without pre-defined criteria for cardiac involvement.
The N-terminal prohormone-B-type natriuretic peptide (NT-proBNP) is a prognostic biomarker of cardiac dysfunction. NT-proBNP baseline values (geometric mean) were 273 nanogram/L and 531 nanogram/L in Amvuttra-treated and placebo-treated patients, respectively. At Month 18, the geometric mean NT-proBNP levels decreased by 6% in Amvuttra patients, while there was a 96% increase in placebo patients.
Centrally-assessed echocardiograms showed changes in LV wall thickness (LS mean difference: -0.18 mm [95% CI -0.74, 0.38]) and longitudinal strain (LS mean difference: -0.4% [95% CI -1.2, 0.4]) with Amvuttra treatment relative to placebo.
Despite the observed values for NT-proBNP and LV wall thickness, a clinical benefit in regard to cardiomyopathy is yet to be confirmed.
5.2 Pharmacokinetic Properties
The pharmacokinetic properties of Amvuttra were characterised by measuring the plasma and urine concentrations of vutrisiran.
Absorption.
Following subcutaneous administration, vutrisiran is rapidly absorbed with a time to maximum plasma concentration (Tmax) of 3.0 (range: 2.0 to 6.5) hours. At the recommended dosing regimen of 25 mg once every 3 months subcutaneously, the mean (% coefficient of variation [%CV]) steady state peak concentrations (Cmax), and area under the concentration time curve from 0 to 24 hours (AUC0-24) were 0.12 microgram/mL (64.3%), and 0.80 microgram.h/mL (35.0%), respectively. There was no accumulation of vutrisiran in plasma after repeated quarterly dosing.
Distribution.
Vutrisiran is greater than 80% bound to plasma proteins over the concentration range observed in humans at the dose of 25 mg once every 3 months subcutaneously. The population estimate for the apparent central compartment volume of distribution (Vd/F) of vutrisiran in humans was 10.2 L (% Relative standard error [RSE] = 5.71%). Vutrisiran distributes primarily to the liver after subcutaneous dosing.
Metabolism.
Vutrisiran is metabolised by endo- and exo-nucleases to short nucleotide fragments of varying sizes within the liver. There were no major circulating metabolites in humans. In vitro studies indicate that vutrisiran does not undergo metabolism by CYP450 enzymes.
Excretion.
Following a 25 mg single subcutaneous dose, the median apparent plasma clearance was 21.4 (range: 19.8, 30.0) L/h. The median terminal elimination half-life (t1/2) of vutrisiran was 5.23 (range: 2.24, 6.36) hours. After a single subcutaneous dose of 5 to 300 mg, the mean fraction of unchanged active substance eliminated in urine ranged from 15.4 to 25.4% and the mean renal clearance ranged from 4.45 to 5.74 L/h for vutrisiran.
Linearity/non-linearity.
Following single subcutaneous doses over the 5 to 300 mg dose range, vutrisiran Cmax was shown to be dose proportional while area under the concentration-time curve from the time of dosing extrapolated to infinity (AUCinf) and area under the concentration-time curve from the time of dosing to the last measurable concentration (AUClast) were slightly more than dose proportional.
Pharmacokinetic/pharmacodynamic relationship(s).
Population pharmacokinetic/pharmacodynamic analyses in healthy subjects and patients with hATTR amyloidosis (n=202) showed a dose-dependent relationship between predicted vutrisiran liver concentrations and reductions in serum TTR. The model-predicted median steady state peak, trough, and average TTR reductions were 88%, 86%, and 87%, respectively, confirming minimal peak-to-trough variability across the 3-month dosing interval. Covariate analysis indicated similar TTR reduction in patients with mild-to-moderate renal impairment or mild hepatic impairment, as well as by sex, race, prior use of TTR stabilisers, genotype (V30M or non-V30M), age and weight.
Special populations.
Gender and race.
Clinical studies did not identify significant differences in steady state pharmacokinetic parameters or TTR reduction according to gender or race.
Elderly patients.
In the HELIOS-A study, 46 (38%) patients treated with vutrisiran were ≥ 65 years old and of these 7 (5.7%) patients were ≥ 75 years old. There were no significant differences in steady state pharmacokinetic parameters or TTR reduction between patients < 65 years old and ≥ 65 years old.
Hepatic impairment.
Population pharmacokinetic and pharmacodynamic analyses indicated no impact of mild hepatic impairment (total bilirubin ≤ 1 x ULN and AST > 1 x ULN, or total bilirubin > 1.0 to 1.5 x ULN and any AST) on vutrisiran exposure or TTR reduction compared to patients with normal hepatic function. Vutrisiran has not been studied in patients with moderate or severe hepatic impairment.
Renal impairment.
Population pharmacokinetic and pharmacodynamic analyses indicated no impact of mild or moderate renal impairment (eGFR ≥ 30 to < 90 mL/min/1.73 m2) on vutrisiran exposure or TTR reduction compared to subjects with normal renal function. Vutrisiran has not been studied in patients with severe renal impairment or end-stage renal disease.
5.3 Preclinical Safety Data
Genotoxicity.
No mutagenic or clastogenic potential of vutrisiran was found in a battery of tests, including a bacterial mutagenicity assay, in vitro chromosomal aberration assay in human lymphocytes, and an in vivo rat bone marrow micronucleus assay.
Carcinogenicity.
Carcinogenicity studies have not been completed.6 Pharmaceutical Particulars
6.1 List of Excipients
Monobasic sodium phosphate dihydrate, dibasic sodium phosphate dihydrate, sodium chloride, water for injections, sodium hydroxide (for pH adjustment), phosphoric acid (for pH adjustment).
6.2 Incompatibilities
In the absence of compatibility studies, this medicinal product must not be mixed with other medicinal products.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 30°C. Do not freeze.
6.5 Nature and Contents of Container
Pre-filled syringe (Type I glass with a bromobutyl rubber, fluoropolymer coated, plunger stopper) with stainless steel 29-gauge needle with a needle shield.
The syringe components are not made with natural rubber latex.
Amvuttra is available in packs containing one single-use pre-filled syringe.
6.6 Special Precautions for Disposal
Each pre-filled syringe is intended for single use in a single patient only.
In Australia, any unused medicine or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
pH: approximately 7.
Osmolality: 210 to 390 mOsm/kg.
Chemical structure.
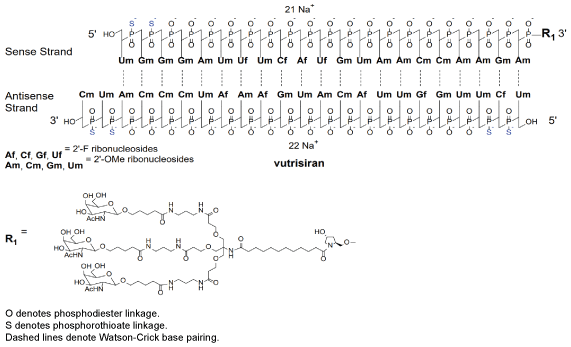
CAS number.
1867157-35-4.7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Summary Table of Changes
