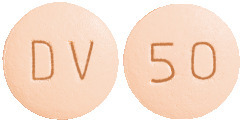
What is in this leaflet
This leaflet answers some common questions about this medicine. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking this medicine against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What this medicine is used for
The name of your medicine is APO-Desvenlafaxine MR Tablets. It contains the active ingredient desvenlafaxine (as benzoate).
It is used in the treatment and prevention of relapse of depression. Depression can affect your whole body and may cause emotional and physical symptoms such as feeling low in spirit, being unable to enjoy life, poor appetite or overeating, disturbed sleep, loss of sex drive, lack of energy and feeling guilty over nothing.
Desvenlafaxine belongs to a class of medications called Serotonin-Noradrenaline Reuptake Inhibitors (SNRIs).
Serotonin and noradrenaline are chemical messengers that allow certain nerves in the brain to work.
This medicine increases the level of these two messengers. Experts think this is how it helps to restore your feeling of wellness.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed this medicine for another reason.
This medicine is available only with a doctor's prescription.
There is no evidence that this medicine is addictive.
This medicine should not be used in children or adolescents under 18 years of age. The safety and effectiveness of this medicine in this age group have not been established.
Before you take this medicine
When you must not take it
Do not take this medicine if you have an allergy to:
- any medicine containing desvenlafaxine or venlafaxine
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not take this medicine if you are taking other medications for depression known as monoamine oxidase inhibitors (MAOIs). Do not take desvenlafaxine even if you have stopped taking any MAOIs but have taken them within the last 14 days.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- a history of fits (seizures or convulsions)
- a personal history or family history of bipolar disorder
- blood pressure problems
- glaucoma (increased pressure in the eye)
- a tendency to bleed more than normal or you are taking a blood thinning medication
- raised cholesterol or lipid levels
- problems with your kidneys or liver
- problems with your heart
- low sodium levels in your blood
- or any other medical conditions.
Tell your doctor if you are currently pregnant or you plan to become pregnant. Do not take this medicine whilst pregnant until you and your doctor have discussed the risks and benefits involved.
One of these risks is that newborn babies whose mothers have been taking this medicine may have several problems including breathing difficulties, seizures, lack of oxygen in their blood, physical and/or behavioural problems, vomiting and diarrhoea.
If you take desvenlafaxine or similar antidepressants mid to late in your pregnancy, you may develop a condition known as "pre-eclampsia", which is characterised by persistent high-blood pressure during or after pregnancy. Symptoms of preeclampsia can include headaches, abdominal pain, shortness of breath or burning behind the sternum, nausea and vomiting, confusion, heightened state of anxiety, and/or visual disturbances such as oversensitivity to light, blurred vision, or seeing flashing spots or auras.
If you take desvenlafaxine or similar antidepressants in the last month of your pregnancy, you may experience heavy bleeding during and/or after delivery.
Continuing treatment with desvenlafaxine or similar antidepressants during pregnancy should be strictly as directed by your doctor. Symptoms of a relapse may occur if treatment is discontinued, even if major depression was previously under control.
Tell your doctor if you are currently breastfeeding, or you plan to breastfeed. This medicine passes into breast milk and there is a possibility that the breastfed baby may be affected.
Do not take this medicine whilst breastfeeding until you and your doctor have discussed the risks and benefits involved.
If you have not told your doctor about any of the above, tell them before you start taking this medicine.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and this one may interfere with each other. These include:
- medications for depression known as monoamine oxidase inhibitors (MAOIs) (such as moclobemide, phenelzine and tranylcypromine).
Tell your doctor if you are taking or have stopped taking them within the last 14 days.
Ask your doctor or pharmacist if you are unsure if you are taking any of these medicines.
It is important that you do not take this medicine or medicines similar to desvenlafaxine with MAOIs or within 14 days of taking a MAOI, as this may result in a serious life-threatening condition. - any other medications for bipolar disorder, depression, anxiety, obsessive-compulsive disorder or pre-menstrual dysphoric disorder, including St John's wort
- drugs that affect serotonin levels e.g. tramadol, dextromethorphan, fentanyl, methadone, amphetamines and pentazocine
- medicines for weight loss, including sibutramine
- medicines used to treat Attention Deficit Hyperactivity Disorder (ADHD) such as dexamphetamine and lisdexamphetamine
- triptans (used to treat migraine)
- linezolid (used to treat infections)
- drugs that affect your tendency to bleed, e.g., aspirin, NSAIDs, warfarin.
- Opiods (used to manage pain)
These medicines may be affected by desvenlafaxine or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
Switching to this medicine from other antidepressants:
Side effects from discontinuing antidepressant medication may occur if you are switched from other antidepressants, including venlafaxine, to this medicine. Your doctor may gradually reduce the dose of your initial antidepressant medication to help reduce these side effects.
How to take this medicine
Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
If you do not understand the directions, ask your doctor or pharmacist for help.
How much to take
Your doctor will tell you how much of this medicine you should take. This will depend on your condition and whether you are taking any other medicines.
The usual dose is 50 mg taken once daily with or without food.
If you have kidney problems, you may need a lower dose of desvenlafaxine.
How to take it
Swallow the tablets whole with a full glass of water.
Do not divide, crush, chew or place the tablets in water.
Do not be concerned if you see a tablet 'shell' in your faeces after taking this medicine. As the tablet travels the length of your gastrointestinal tract, the active ingredient desvenlafaxine is slowly released. The tablet 'shell' remains undissolved and is eliminated in your faeces. Therefore, even though, you may see a tablet 'shell' in your faeces, your dose of desvenlafaxine has been absorbed.
When to take it
Take your medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
It does not matter if you take this medicine before or after food.
How long to take it for
Continue taking your medicine for as long as your doctor tells you.
Although you may begin to feel better after two weeks, it may take several weeks before you feel much better. It is important to give this medicine time to work.
This medicine helps to control your condition but does not cure it, so it is important to keep taking your medicine even if you feel well.
Make sure you have enough to last over weekends and holidays.
If you forget to take it
If it is less than 12 hours until your next dose, skip the missed dose and take your next dose at the usual time.
Otherwise, take it as soon as you remember and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for missed doses. This may increase the chance of you experiencing side effects.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints to help you remember.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much of this medicine. Do this even if there are no signs of discomfort or poisoning.
You may need urgent medical attention.
While you are taking this medicine
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking this medicine.
Tell any other doctors, dentists, and pharmacists who treat you that you are taking this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are taking this medicine. It may affect other medicines used during surgery.
If you become pregnant or start to breastfeed while taking this medicine, tell your doctor immediately.
If you are about to have any blood tests, tell your doctor that you are taking this medicine. It may interfere with the results of some tests.
Keep all your doctor's appointments so that your progress can be checked. Your doctor may do some tests from time to time to make sure the medicine is working and to prevent unwanted side effects.
Always discuss any questions you have about this medicine with your doctor.
Take this medicine exactly as your doctor has prescribed.
Watch carefully for signs that your depression is getting worse, especially in the first few weeks of treatment or if your dose has changed. Sometimes people with depression can experience a worsening of their depressive symptoms. This can happen even when taking an antidepressant.
Tell your doctor there is the potential for a false positive urinary drug screen while on this medicine.
Tell your doctor immediately if you experience any of the following symptoms, especially if they are severe, you have not had these symptoms before, or they happen very suddenly:
- anxiety or agitation
- panic attacks
- difficulty sleeping
- irritability
- aggressiveness
- hostility or impulsiveness
- restlessness
- overactivity or uninhibited behaviour
- other unusual changes in behaviour
- thoughts of suicide.
Tell your doctor immediately if you have any thoughts about suicide or doing harm to yourself.
Warning signs of suicide
If you or someone you know is showing the following warning signs, contact your doctor or a mental health advisor right away or go to the nearest hospital for treatment.
All thoughts or talk about suicide or violence are serious. These include:
- thoughts or talk about death or suicide
- thoughts or talk about self-harm or doing harm to others
- any recent attempts of self-harm
- an increase in aggressive behaviour, irritability or agitation.
Things you must not do
Do not take this medicine to treat any other complaints unless your doctor tells you to.
Do not give your medicine to anyone else, even if they have the same condition as you.
Do not stop taking your medicine or lower the dosage without checking with your doctor. Your doctor may want to slowly decrease your dose of this medicine to help avoid side effects. Side effects are known to occur when people stop taking this medicine, especially when they suddenly stop therapy.
Some of these side effects include:
- headache
- nausea
- dizziness
- tiredness
- irritability
- anxiety
- abnormal dreams
- diarrhoea
- excessive sweating
- visual impairment
- high blood pressure
Slowly reducing the amount of this medicine being taken reduces the possibility of these effects occurring. In some people this may need to occur over periods of months or longer.
Things to be careful of
Be careful when driving or operating machinery until you know how this medicine affects you. It may make you feel drowsy.
Avoid drinking alcohol while you are taking this medicine.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking this medicine.
All medicines can have side effects. Sometimes they are serious but most of the time they are not.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor or pharmacist if you notice any of the following and they worry you:
- nausea or vomiting
- loss of appetite
- diarrhoea
- constipation
- difficulty passing urine
- difficulty sleeping, abnormal sleepiness or abnormal dreams
- sexual function problems such as decreased sex drive, delayed ejaculation, problems achieving erection or difficulties achieving orgasm – sometimes lasting even after you have stopped taking desvenlafaxine
- nervousness or anxiety
- feeling jittery or irritable
- difficulty thinking or working
- yawning
- disturbances in concentration
- fainting or dizziness after standing up
- fatigue
- rapid heart beat
- chills
- headache
- excessive sweating
- hot flushes
- rash
- weight loss or weight gain
- blurred vision
- ringing in the ears
- altered taste, dry mouth.
Tell your doctor as soon as possible if you notice any of the following:
- muscle spasms, stiffness, weakness or movement disorders
- abnormal facial movements such as tongue thrusting, repetitive chewing, jaw swinging, or grimacing
- a feeling of apathy or not caring about things
- feeling detached from yourself
- hallucinations, confusion
- agitation
- unusually overactive
- problems with breathing, shortness of breath
- bleeding or bruising more easily than normal
- numbness or pins and needles
- sensitivity to sunlight.
The above list includes serious side effects that may require medical attention. Serious side effects are rare.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- palpitations, shortness of breath, intense chest pain, or irregular heartbeats
- severe upper abdominal pain
- swollen and tender abdomen
- fever
- rise or decrease in blood pressure – you may experience headache, blurred vision, palpitations, confusion or loss of consciousness, or sometimes you may not experience any of these symptoms. It is important to keep your routine doctor's appointments so that your blood pressure can be checked seizures or fits.
- symptoms of sudden fever with sweating, rapid heartbeat and muscle stiffness, which may lead to loss of consciousness
- symptoms of an allergic reaction including cough, shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue, throat or other parts of the body; rash, itching or hives on the skin.
The above list includes very serious side effects. You may need urgent medical attention or hospitalisation. These side effects are very rare.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may occur in some patients.
Some of these side effects (for example, increase in blood pressure, increase in blood cholesterol, changes to liver function, protein in the urine) can only be found when your doctor does tests from time to time to check your progress.
Storage and disposal
Storage
Keep your medicine in its original packaging until it is time to take it. If you take your medicine out of its original packaging it may not keep well.
Keep your medicine in a cool dry place where the temperature will stay below 25°C.
Do not store your medicine, or any other medicine, in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep this medicine where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What APO-Desvenlafaxine MR Tablet looks like
50 mg Tablet
Light pink, biconvex, round shaped, film coated tablets debossed with "DV" on one side and "50" on the other side. AUST R 218307.
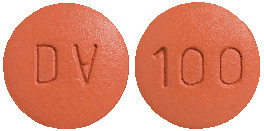
100 mg Tablet
Reddish-orange, biconvex, round shaped, film coated tablets, debossed with "DV" on one side and "100" on the other side. AUST R 227802.
Available in blister packs of 7, 14 and 28 tablets.
* Not all strengths and/or pack sizes may be available.
Ingredients
Each modified release tablet contains 50 mg or 100 mg of desvenlafaxine benzoate as the active ingredient.
It also contains the following inactive ingredients:
- Microcrystalline cellulose,
- purified talc,
- stearic acid,
- colloidal anhydrous silica,
- magnesium stearate,
- hypromellose
- OPADRY® II complete film coating system 85F94487 PINK (PI No. 106952) for 50 mg tablets
- OPADRY® II complete film coating system 85F94527 PINK (PI No. 106953) for 100 mg tablet
This medicine is gluten-free, lactose-free, sucrose-free, tartrazine-free and free of other azo dyes.
Sponsor
Arrotex Pharmaceuticals Pty Ltd
15 – 17 Chapel Street,
Cremorne, Victoria 3121
http://arrotex.com.au
This leaflet was last updated in November 2022.
Published by MIMS May 2025

 Molecular Formula: C23H31NO4 (as benzoate).
Molecular Formula: C23H31NO4 (as benzoate).