


What is in this leaflet
This leaflet answers some common questions about topiramate. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using this medicine against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may want to read it again.
What this medicine is used for
Topiramate is used to:
- treat various types of epilepsy in adults and children over 2 years of age
- prevent migraines in adults - it is not used to treat severe migraines that come on suddenly (acute)
Topiramate belongs to a group of medicines called antiepileptic drugs.
How it works
Topiramate prevents seizures and migraines by acting on the nerves and chemicals in the brain.
Your doctor may prescribe topiramate on its own, or in addition to another medicine for controlling your seizures or migraines.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed this medicine for another reason.
This medicine is available only with a doctor's prescription.
This medicine is not addictive.
This medicine may be used to treat epilepsy in children aged 2 years or older.
Before you take this medicine
When you must not take it
Do not take this medicine if you have an allergy to:
- topiramate
- any of the ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction may include:
- shortness of breath, wheezing or difficulty breathing
- swelling of the face, lips, tongue, throat or other parts of the body
- rash, itching or hives on the skin
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn, shows signs of tampering or it does not look quite right. If it has expired or is damaged, return it to your pharmacist for disposal.
Do not breastfeed if you are taking this medicine.
Before you start to take it
You must tell your doctor if you:
- are pregnant or planning to become pregnant. Topiramate has caused harm to the developing foetus when administered to a pregnant woman. Its safety has not been verified in pregnant humans. Information available indicates that there is an association between the use of this medicine in humans during pregnancy and birth defects including cleft lip/ palate, and neurodevelopmental disorders such as autism spectrum disorders and intellectual disability. However, it is very important to control your fits while you are pregnant. If it is necessary for you to take this medicine, your doctor can help you decide whether or not to take it during pregnancy
- are breast feeding or wish to breastfeed. Topiramate may appear in breast milk and it is not recommended to breastfeed while taking Topiramate
- have or have had personality disorder or mental illness
- have or have ever had kidney stones, kidney disease or have a family history of kidney stones
- have or have every had liver disease
- have eye problems or high pressure in the eye
- history of metabolic acidosis (too much acid in the blood, which may cause an increased rate of breathing)
If you have not told your doctor about any of the above, tell them before you start taking this medicine.
Your doctor will advise you whether or not to take this medicine or if you need to adjust the dose or adapt your treatment.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
In particular, tell your doctor or pharmacist if you are taking:
- other medicines for epilepsy (e.g. phenytoin, carbamazepine)
- digoxin (used to treat heart problems)
- contraceptives such as estrogen containing or progestin only contraceptives
- metformin, pioglitazone or glibenclamide (used to treat diabetes)
- lithium or risperidone (used to treat bipolar disorder and schizophrenia)
- haloperidol (used to treat psychoses)
- hydrochlorothiazide (used to treat swelling or high blood pressure)
- propanol (used in high blood pressure, some heart conditions, tremors, tumor or migraine headaches)
- diltiazem (used to treat hypertension and prevent angina)
- valproic acid (used to treat epilepsy or mood disorders)
- vitamin K-antagonist anticoagulant medications (such as warfarin)
- amitriptyline (used to treat depression)
- flunarizine
- any medicine which slows your reactions (CNS depressants). This may include medicines to help you sleep or relieve pain, antidepressants, tranquillisers or antihistamines which can make you drowsy.
These medicines may be affected by topiramate or may affect how well it works.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How to take this medicine
Follow all directions given to you by your doctor carefully. They may differ to the information contained in this leaflet.
If you do not understand the instructions on the pack, ask your doctor or pharmacist for help.
How much to take
Your doctor will tell you how much of this medicine you should take. This will depend on your condition and whether you are taking any other medicines.
The doses shown below are the usual recommended doses. However, your doctor may tell you to take higher or lower doses.
Adults – Epilepsy
The usual starting dose is 25 mg to 50 mg taken at night for one week or longer. The dose can be gradually increased by 25 mg to 100 mg over weekly periods or longer, until a suitable dose is reached.
Adults – Migraine
The usual starting dose is 25 mg taken at night for one week. The dose can be gradually increased by 25mg over weekly periods or longer, until a suitable dose is reached.
Children (2 years and over) - Epilepsy
The usual starting dose is 25 mg or less per day, depending on the child's body weight. The dose is then gradually increased over weekly periods or longer, until a suitable dose is reached.
How to take it
Swallow the tablets whole with a glass of water. Do not crush or chew the tablets.
When to take it
Take this medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
It does not matter if you take it with or without food.
How long to take it for
Continue taking your medicine for as long as your doctor tells you.
This medicine helps to control your condition but does not cure it. It is important to keep taking your medicine even if you feel well.
If you forget to take it
If it is almost time to take your next dose, skip the dose you missed and take your next dose when you are meant to.
Otherwise, take it as soon as you remember and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for missed doses. This may increase the chance of you experiencing side effects.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice or go to Accident and Emergency at your nearest hospital if you think that you or anyone else may have taken too much of this medicine.
Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
If you take too much topiramate you may experience headache, dizziness, light-headedness, drowsiness, convulsions, speech disturbances, double or blurred vision, difficulty with thinking, abnormal coordination, stomach pain, depression, agitation, faster breathing or you may lose consciousness.
While you are taking this medicine
Things you must do
Drink plenty of water. Topiramate has been known to cause kidney stones and drinking water may help prevent this.
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are taking this medicine.
Tell any other doctors, dentists and pharmacists who are treating you that you take this medicine.
If you become pregnant or start to breastfeed while taking this medicine, tell your doctor immediately.
Tell your doctor if you are going to have surgery or an anaesthetic or are going into hospital.
Keep all of your doctor's appointments so that your progress can be checked. Your doctor may occasionally do tests to make sure the medicine is working and to prevent side effects.
Things you must not do
Do not give this medicine to anyone else, even if they have the same condition as you.
Do not take your medicine to treat any other complaint unless your doctor tells you to.
Do not drink alcohol.
Do not drive or operate machinery until you know how this medicine affects you.
Do not stop taking your medicine or change the dosage without first checking with your doctor. If you stop taking topiramate for seizures abruptly, your epilepsy may worsen or come back (this is known as 'rebound seizures').
Your doctor will advise you if you need to stop taking topiramate and, if so, how to do this safely. Talk to your doctor if you are unsure whether you should stop taking topiramate.
Things to be careful of
Changes to your medicine.
If you are seizure free or your seizures are well controlled, a reduction in your dose, discontinuation or substitution of your current medication should first be assessed by your doctor and pharmacist, and any changes should be implemented gradually.
Effects on thoughts and behaviour
Medicines used to treat epilepsy can increase the risk of suicidal thoughts and behaviour. If you experience feelings of deep sadness and unworthiness (depression) or a worsening of these feelings, any unusual changes in your mood or the emergence of suicidal thoughts, behaviour or thoughts of self-harm, you should report this to your doctor immediately.
Decreased sweating and elevation in body temperature
Topiramate may cause decreased sweating and increased body temperature (fever). People, especially children, should be watched for signs of decreased sweating and fever, especially in hot temperatures. Some people may need to be hospitalized for this condition. Call your healthcare provider right away if you have a high fever, a fever that does not go away, or decreased sweating.
Effects on driving and operating machinery
Topiramate may cause drowsiness, dizziness or other symptoms which could affect your ability to drive or operate machinery. It may also cause visual disturbances or blurred vision. Make sure you know how you are affected by this medicine before you drive or use machinery.
Particular care is recommended when you first start taking this medicine or if your dose of topiramate or any other medicine changes.
Effects of food and alcohol
Topiramate can be taken with or without food.
Do not drink alcohol while taking this medicine. Alcohol may increase the risk of you experiencing side effects, such as drowsiness.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking topiramate.
This medicine helps most people, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice any of the following:
- dizziness
- decrease in appetite or weight loss
- itchy skin or skin rash
- inability to sleep
- tingling and numbness of hands and feet (pins and needles)
- nausea, diarrhoea, vomiting or constipation
- abdominal pain or discomfort
- ear pain, buzzing or ringing in ears, deafness
Tell your doctor as soon as possible if you notice any of the following as you may need medical attention:
- depression
- nervousness or feeling anxious
- mood alterations such as aggression, agitation or anger
- disturbance in attention
- difficulty with memory or memory impairment
- slowing of thought processes
- abnormal behaviour
- expressive language disorder
- difficulty in speaking
- balance disorder
- co-ordination problems
- unusual hair loss or thinning
- abnormal frequent urination
- decreased feeling or sensitivity, especially in the skin
- fever or high temperature
- decreased or lack of sweating or overheating (mainly in children)
- unusual weakness
- taste disturbance or loss of taste
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- unusual tiredness, drowsiness, irritability, or lack of energy
- difficulty breathing, fast or irregular heartbeat or tightening of chest
- thoughts of harming yourself or thoughts of suicide
- kidney stones
- pain when passing urine
- sudden changes in your eyesight (e.g. blurred or loss of vision) or rapid uncontrollable movements of the eyes
- eye pain or increased pressure in the eye
- unexplained bleeding or bleeding more frequently
- severe blisters and bleeding in mucosal sites (such as lips, eyes, mouth, nose, genitals) that may cause the skin to peel, sometimes occurring with fever and flu-like symptoms
These are serious side effects. You may need urgent medical attention. Serious side effects are rare.
Other side effects not listed above may also occur in some people. Tell your doctor if you notice any other effects to time to check your progress.
Storage and disposal
Storage
Keep your medicine in the pack until it is time to take it. If you take your medicine out of the pack it may not keep well.
Keep your medicine in a cool dry place where the temperature stays below 25°C.
Do not store your medicine or any other medicine in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep this medicine where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine that is left over.
Product description
What it looks like
The tablets are available in 4 different strengths:
25 mg tablets:
White to off-white, round, unscored, film coated tablet, imprinted "APO" on one side and "TP over "25" on the other side.
Blister pack of 60 tablets.
AUST R 124729.
Bottle of 60 tablets.
AUST R 124733.
50 mg tablets:
Light-yellow, round, unscored, film coated tablet, imprinted "APO" on one side and "TP over "50" on the other side.
Blister pack of 60 tablets.
AUST R 124730.
Bottle of 60 tablets.
AUST R 124734.
100 mg tablets:
Mustard yellow, round, unscored, film coated tablet, imprinted "APO" on one side and "TP over "100" on the other side.
Blister pack of 60 tablets.
AUST R 124731.
Bottle of 60 tablets.
AUST R 124735.
200 mg tablets:
Reddish-brown, round, unscored, film coated tablet, imprinted "APO" on one side and "TP over "200" on the other side.
Blister pack of 60 tablets.
AUST R 124732.
Bottle of 60 tablets.
AUST R 124736.
* Not all strengths, pack types and/or pack sizes may be available.
Ingredients
Each tablet contains 25 mg, 50 mg, 100 mg or 200 mg of topiramate as the active ingredient.
It also contains the following:
- methylcellulose
- croscarmellose sodium
- magnesium stearate
- colloidal anhydrous silica
- hypromellose
- hyprolose
- macrogol
- titanium dioxide
- iron oxide yellow (50 mg & 100 mg only)
- iron oxide red (200 mg only).
This medicine does not contain gluten, lactose, sucrose, tartrazine or any other azo dyes.
Sponsor
Arrotex Pharmaceuticals Pty Ltd
15 - 17 Chapel Street
Cremorne, VIC 3121
Australia
Web: www.arrotex.com.au
Tel: +61-1300927769
This leaflet was last updated in August 2023.
Published by MIMS October 2023
 It is not necessary to monitor topiramate plasma concentrations to optimise topiramate therapy. For patients receiving concomitant phenytoin and carbamazepine, dosage adjustment for topiramate may be required (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
It is not necessary to monitor topiramate plasma concentrations to optimise topiramate therapy. For patients receiving concomitant phenytoin and carbamazepine, dosage adjustment for topiramate may be required (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions). The relative risk for suicidal thoughts or behaviour was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.
The relative risk for suicidal thoughts or behaviour was higher in clinical trials for epilepsy than in clinical trials for psychiatric or other conditions, but the absolute risk differences were similar for the epilepsy and psychiatric indications.




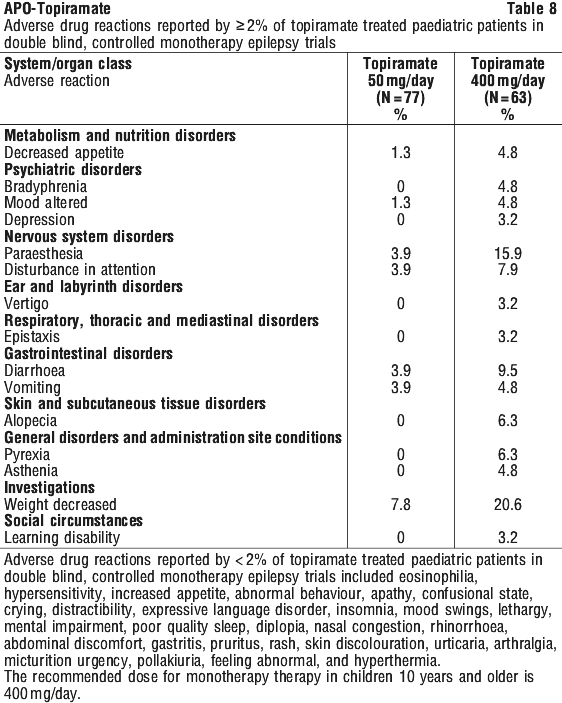

 ADRs reported, rate unspecified, in open label clinical trials of topiramate treated pediatric patients are shown in Table 11.
ADRs reported, rate unspecified, in open label clinical trials of topiramate treated pediatric patients are shown in Table 11.

 The overall safety profile of topiramate observed in the migraine studies was generally consistent with that established for epilepsy therapy.
The overall safety profile of topiramate observed in the migraine studies was generally consistent with that established for epilepsy therapy. Chemical name: 2,3:4,5-bis-0-(1-methylethylidene)-β-D-fructopyranose sulfamate.
Chemical name: 2,3:4,5-bis-0-(1-methylethylidene)-β-D-fructopyranose sulfamate.