

What is in this leaflet
This leaflet answers some common questions about venlafaxine. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using this medicine against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may want to read it again.
What this medicine is used for
Venlafaxine is used to treat major depression, and to prevent it coming back. It is also used to treat panic attacks and anxiety.
Venlafaxine belongs to a group of medicines called serotonin-noradrenaline reuptake inhibitors (SNRIs).
Venlafaxine increases the level of serotonin and noradrenaline in the brain, helping to restore your feeling of wellness.
Depression may cause emotional and physical symptoms such as feeling low, poor appetite or lack of energy.
Excessive anxiety may cause you to feel constantly and uncontrollably worried and distressed. It may also make you feel irritable, and cause difficulty in thinking and sleeping.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed Venlafaxine XR for another reason.
This medicine is available only with a doctor's prescription.
This medicine is not addictive.
There is not enough information to recommend the use of this medicine for children under the age of 18 years.
Before you take this medicine
When you must not take it
Do not take this medicine if you have an allergy to:
- venlafaxine
- any of the ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue, throat or other parts of the body
- rash, itching or hives on the skin
Do not take this medicine if you are taking other medicines called Monoamine Oxidase Inhibitors (MAOIs).
MAOIs may be used for the treatment of depression (phenelzine, tranylcypromine or moclobemide), Parkinson's disease (selegiline), infections (linezolid) or diagnosis of certain conditions/treatment of certain blood disorders (methylene blue).
Taking venlafaxine with a MAOI may cause a serious reaction with a sudden increase in body temperature, extremely high blood pressure and severe convulsions.
Do not take venlafaxine until 14 days after stopping most MAOIs, and do not take any MAOIs until more than one week after stopping venlafaxine. You may take venlafaxine 24 hours after stopping the reversible MAOI called moclobemide.
Do not take this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, return it to your pharmacist for disposal.
If you are not sure whether you should start taking this medicine, talk to your doctor.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- epilepsy, fits or seizures
- a personal or family history of bipolar disorder
- a history of aggression
- a history of restlessness or difficulty sitting still
- drug abuse or misuse
- blood pressure problems
- diabetes
- acute angle glaucoma or increased pressure in the eye
- a tendency to bleed more than normal or other blood disorders
- raised cholesterol levels or you are taking medicines to lower cholesterol
- kidney or liver problems
- low sodium levels in your blood, or a condition called SIADH (Syndrome of Inappropriate Antidiuretic Hormone Secretion)
- thoughts or actions relating to self-harm or suicide
- heart problems, especially conditions causing irregular heartbeats – your doctor may wish to do some heart tests such as an electrocardiogram (ECG) or blood tests during treatment with venlafaxine
Tell your doctor if you are pregnant or plan to become pregnant. There have been reports that babies exposed to venlafaxine and other antidepressants during the third trimester of pregnancy may develop complications after birth, including breathing difficulties, seizures and lack of oxygen in their blood.
Continuing treatment with venlafaxine or similar antidepressants during pregnancy should be strictly as directed by your doctor.
Do not take this medicine whilst pregnant until you and your doctor have discussed the risks and benefits involved.
Tell your doctor if you are breastfeeding. Venlafaxine passes into breast milk and there is a possibility that your baby may be affected. For this reason, the use of venlafaxine is not recommended if you are breastfeeding.
If you have not told your doctor about any of the above, tell them before you start taking this medicine.
Taking other medicines
Tell your doctor and pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines may interact with venlafaxine. These include:
- MAOIs (e.g. phenelzine, tranylcypromine, moclobemide, selegiline, linezolid or methylene blue).
Do not take venlafaxine until 14 days after stopping most MAOIs, and do not take any MAOIs until more than one week after stopping venlafaxine.
For moclobemide, do not take venlafaxine until at least 24 hours after stopping moclobemide, and do not take moclobemide until more than one week after stopping venlafaxine. - St John's wort (Hypericum perforatum) or tryptophan
- dopamine agonists used to treat Parkinson's disease
- any other medications for depression, anxiety, obsessive-compulsive disorder or premenstrual dysphoric disorder
- haloperidol, risperidone, lithium or clozapine, used to treat mood disorders
- tramadol, fentanyl, tapentadol, pethidine and methadone, strong pain killers
- amphetamines, including those used to treat Attention Deficit Hyperactivity Disorder (e.g. dexamphetamine and lisdexamphetamine)
- cimetidine, used to treat reflux
- triptans, used to treat migraines
- amiodarone or quinidine, used to treat irregular heartbeats
- warfarin and clopidogrel, used to prevent blood clots
- diuretics, also called fluid or water tablets
- medicines used to treat diabetes
Your doctor may also wish to do some heart tests such as an electrocardiogram (ECG) or blood tests if you are using any of the following whilst taking venlafaxine:
- warfarin, used to prevent blood clotting
- indinavir, an antiviral medicine
- linezolid or erythromycin, used to treat infections
- ketoconazole or fluconazole, used as antifungal medicines
- medicines used for weight loss (e.g. phentermine, sibutramine)
- metoprolol, used to treat high blood pressure or angina
- medications that can affect your heart beat
- grapefruit juice
These medicines may be affected by venlafaxine or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
Other interactions not listed above may also occur.
How to take this medicine
Follow all directions given to you by your doctor or pharmacist carefully. They may differ to the information contained in this leaflet.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
How much to take
Your doctor will tell you how much of this medicine you should take, depending on your condition and if you are taking any other medicines.
The usual starting dose is 75 mg taken once daily.
If necessary, after two weeks, your doctor may increase your dose. The maximum dose you should take in one day is 225 mg.
How to take it
Swallow the capsules whole with a glass of water. Do not divide, crush, chew or place the capsules in water.
Inside venlafaxine XR capsules, there are tablets that contain the venlafaxine. As the medicine travels the length of your gastrointestinal tract, venlafaxine is slowly released.
When to take it
Take this medicine at about the same time each day. Taking it at the same time each day will have the best effect. It will also help you remember when to take it.
How long to take it for
Continue taking your medicine for as long as your doctor tells you.
Although you may begin to feel better after two weeks, it may take several weeks before you feel much better.
Even when you feel well again, you may need to keep taking venlafaxine for several months to make sure the benefits last.
Make sure you have enough to last over weekends and holidays.
If you forget to take it
If it is less than 12 hours until your next dose, skip the missed dose and take your next dose at the usual time.
Otherwise take it as soon as you remember and then go back to taking your medicine as you would normally.
Do not take a double dose to make up for missed doses. This may increase the chance of you experiencing side effects.
Contact your doctor if you have missed more than two doses in a row.
If you have trouble remembering to take your medicine, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre (telephone 13 11 26) for advice or go to the Emergency Department at your nearest hospital if you think that you or anyone else may have taken too much of this medicine. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
If you take too much venlafaxine, you may experience sleepiness, vomiting, dilated pupils, increased heart rate, changes in heart rhythm, seizure (fits), breathing difficulties or becoming unconscious.
While you are taking this medicine
Things you must do
Watch carefully for signs that your depression is getting worse, especially in the first few weeks of treatment or if your dose has changed.
Tell your doctor immediately if you experience any of the following symptoms, especially if they are severe, you have not had these symptoms before, or they happen very suddenly:
- anxiety or agitation
- panic attacks
- difficulty sleeping
- restlessness
- irritability
- aggressiveness, hostility or impulsiveness
- overactivity or uninhibited behaviour
- other unusual changes in behaviour
- thoughts of suicide.
Tell your doctor immediately if you have any thoughts about suicide or doing harm to yourself.
All thoughts or talk about suicide or violence are serious. If you or someone you know is showing the following warning signs, either contact your doctor or a mental health advisor right away or go to the nearest hospital for treatment.
- thoughts or talk about death or suicide.
- thoughts or talk about self-harm or doing harm to others.
- any recent attempts of self-harm.
- an increase in aggressive behaviour, irritability or agitation.
If you are being treated for depression, discuss with your doctor any problems you may have and how you feel, especially any feelings of severe sadness, thoughts of suicide, bursts of unusual energy, anger or aggression, or if you become particularly agitated or restless.
If you are about to be started on any new medicine, tell your doctor that you are taking this medicine.
Tell any other doctors, dentists and pharmacists who are treating you that you take this medicine.
If you become pregnant while taking this medicine, tell your doctor immediately. Make sure your midwife and/or doctor know immediately that you are pregnant and taking venlafaxine, as there is a possibility of problems developing in unborn children.
Symptoms such as feeding difficulty, vomiting, tremor, irritability and constant crying have been reported rarely in newborn babies after mothers have taken venlafaxine in the last 3 months of pregnancy.
In addition, when taken particularly in the last 3 months of pregnancy, medicines like venlafaxine may increase the risk of a serious condition in babies, called persistent pulmonary hypertension of the newborn (PPHN), making the baby breathe faster and appear bluish. These symptoms usually begin during the first 24 hours after the baby is born. If this happens to your baby, you should contact your midwife and/or doctor immediately.
Tell your doctor if you are breastfeeding or are planning to breastfeed.
Tell your doctor if you are about to have any blood tests.
Keep all of your doctor's appointments so that your progress can be checked.
Your doctor may occasionally do tests (such as an electrocardiogram (ECG) or blood tests) to make sure the medicine is working and to prevent side effects. Some people (especially older people or those taking diuretics) may experience a lack of sodium in the blood when taking this medicine.
Tell your doctor if you get a headache or start to feel dizzy, confused, forgetful, weak, unsteady or unable to concentrate. Make sure to drink plenty of fluids.
Things you must not do
Do not give this medicine to anyone else, even if they have the same condition as you.
Do not take your medicine to treat any other complaint unless your doctor tells you to
Do not stop taking your medicine, or change the dosage, without first checking with your doctor. Check with your doctor for the best way to slowly reduce the amount of venlafaxine you are taking before stopping completely, as this can reduce the possibility of these effects occurring.
Side effects from stopping treatment with venlafaxine suddenly may include headache, nausea and vomiting, insomnia, nervousness, anxiety, confusion and agitation, diarrhoea, sweating, flu-like symptoms, loss of appetite, dizziness, impaired coordination and balance, visual impairment, increased blood pressure, tremor, tingling or numbness of the hands and feet.
Slowly reducing your venlafaxine dose reduces the possibility of these effects occurring.
Some of these symptoms may impair driving, or the operation of dangerous machinery. Avoid these activities if you experience these symptoms.
Things to be careful of
Be careful driving or operating machinery until you know how this medicine affects you. Venlafaxine may make you feel drowsy.
Avoid drinking alcohol while you are taking venlafaxine.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking venlafaxine.
All medicines can have side effects.Sometimes they are serious but most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Tell your doctor or pharmacist if you notice any of the following:
- stomach problems e.g. nausea, vomiting, loss of appetite, diarrhoea, constipation or wind
- difficulty sleeping or abnormal dreams
- yawning, feeling drowsy or sedated
- paranoia or aggression
- sexual function problems such as delayed ejaculation, erection problems, decreased sex drive or difficulties achieving orgasm (which may continue even after stopping treatment)
- nervousness, teeth grinding
- excessive enthusiasm or desire, delusions (mania)
- fainting or dizziness after standing up
- restlessness, agitation or difficulty sitting still
- headache
- heavy or irregular menstrual periods
- sweating (including night sweats), hot flushes
- rash, itchiness
- hair loss
- weight loss or weight gain
- unusual milk production
- dilated pupils, sensitivity to sunlight
- ringing in the ears
- altered taste, dry mouth, sore throat
- increased blood pressure or blood cholesterol levels
The above list includes the more common side effects. Mostly, these are mild.
Tell your doctor as soon as possible if you notice any of the following:
- blurred vision
- cloudy urine, problems passing urine, passing urine more frequently, or being unable to control urination
- muscle tremors, spasms, twitching, jerky movements or sustained muscle contractions
- impaired coordination and balance
- increase in bone fractures
- abnormal facial movements such as tongue thrusting, repetitive chewing, jaw swinging, or grimacing
- changes in muscle tone, muscle weakness or fatigue
- a feeling of apathy or not caring about things or not being part of your body
- hallucinations, confusion or agitation
- excessively overactive
- rapid heart beat
- problems with breathing, cough
- sensitivity to sunlight
- bleeding or bruising more easily than normal
- numbness, pins and needles
The above list includes serious side effects. You may need medical attention. Most of these side effects are rare.
If you experience any of the following, stop taking your medicine and contact your doctor immediately or go to the Emergency department at your nearest hospital:
- fits or seizures, which may be accompanied by a fever
- symptoms of sudden fever with sweating, rapid heartbeat and muscle stiffness, which may lead to loss of consciousness
- palpitations, fainting, shortness of breath, chest pain or irregular heartbeat
- dark, red or cola coloured urine, muscle weakness and tenderness, stiffness or aching
- stomach pain, yellowing of the skin, nausea, fever, clammy skin and sweating
- yellowing of the skin or eyes, fever, fatigue, loss of appetite, dark coloured urine or lightcoloured bowel movements
- a severe skin reaction with painful red areas and large blisters, accompanied by fever and chills, aching muscles and generally feeling unwell.
- symptoms of high fever, agitation, confusion, trembling and abrupt contractions of muscles
- severe chills, fever, sore throat and mouth ulcers
- black sticky bowel motions or bloody diarrhoea
- shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue, throat or other parts of the body; rash, itching or hives on the skin (signs of an allergic reaction)
The above list includes very serious side effects. These effects are usually very rare. You may need urgent medical attention or hospitalisation.
Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Other side effects not listed above may occur in some patients.
Some of these side effects, such as increases in blood pressure or blood cholesterol levels, can only be found when your doctor does tests from time to time to check your progress.
Other side effects not listed above may occur in some patients.
Storage and disposal
Storage
Keep your medicine in its pack until it is time to take it. If you take your medicine out of its pack it may not keep well.
Keep your medicine in a cool dry place where the temperature will stay below 25°C.
Do not store your medicine, or any other medicine, in the bathroom or near a sink. Do not leave it on a window sill or in the car. Heat and dampness can destroy some medicines.
Keep this medicine where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking this medicine or the expiry date has passed, ask your pharmacist what to do with any medicine left over.
Product description
What it looks like
75 mg capsules
Peach opaque/peach opaque size '1' hard gelatin capsules having thick and thin radial circular bands on the body and cap in red ink. The capsule is filled with white to off white, round, biconvex, film coated mini tablets. AUST R 286958.
150 mg capsules:
Dark orange/dark orange opaque size '0' hard gelatin capsules having thick and thin radial circular bands on the body and cap in white ink. The capsule is filled with white to off white, round, biconvex, film coated mini tablets. AUST R 286957.
Blister pack of 28 capsules.
*Not all strengths may be available.
Ingredients
Each capsule contains 75 or 150 mg of venlafaxine hydrochloride as the active ingredient.
It also contains the following:
- microcrystalline cellulose
- povidone
- ethanol
- purified talc
- colloidal anhydrous silica
- magnesium stearate
- ethylcellulose
- copovidone
- brilliant blue FCF
- allura red AC
- sunset yellow FCF (only for 150 mg)
- iron oxide black (only for 75 mg)
- iron oxide red (only for 75 mg)
- titanium dioxide
- gelatin
- TekPrint SB-0007P White Ink (150 mg capsules only)
- TekPrint SB-1033 Red Ink (75 mg capsules only)
May contain trace amounts of phenylalanine and sulphites.
This medicine does not contain gluten, lactose, sucrose, tartrazine or any other azo dyes.
Sponsor
Arrotex Pharmaceuticals Pty Ltd
15-17 Chapel st
Cremorne VIC 3121
Australia
Tel: 1800 195 055
This leaflet was last updated in August 2023.
Published by MIMS September 2023
 Below are adverse reactions from combined analyses of the clinical studies for major depression, generalised anxiety disorder, social anxiety disorder, and panic disorder. The adverse reactions have been presented using the Council for International Organizations of Medical Sciences (CIOMS) frequency categories: common: ≥ 1%; uncommon: ≥ 0.1% and < 1%; rare: ≥ 0.01% and < 0.1%; very rare: < 0.01%.
Below are adverse reactions from combined analyses of the clinical studies for major depression, generalised anxiety disorder, social anxiety disorder, and panic disorder. The adverse reactions have been presented using the Council for International Organizations of Medical Sciences (CIOMS) frequency categories: common: ≥ 1%; uncommon: ≥ 0.1% and < 1%; rare: ≥ 0.01% and < 0.1%; very rare: < 0.01%.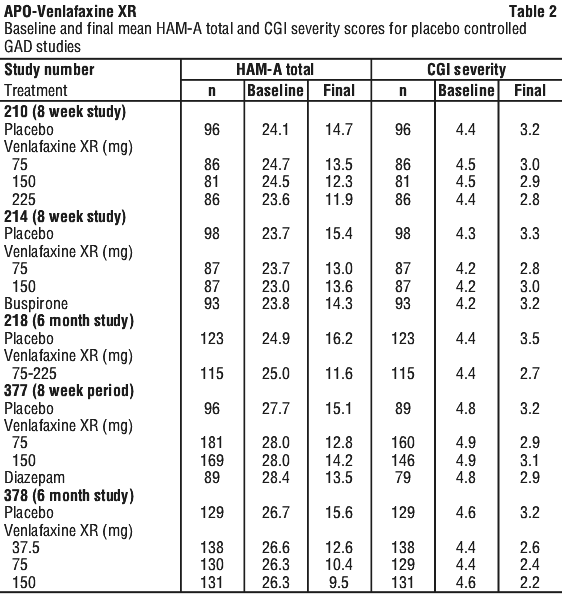

 Examination of subsets of the population studied did not reveal any differential responsiveness on the basis of gender. There was insufficient information to determine the effect of age or race on outcome in these studies.
Examination of subsets of the population studied did not reveal any differential responsiveness on the basis of gender. There was insufficient information to determine the effect of age or race on outcome in these studies.
 Chemical name: 1-[(1RS)-2-(Dimethylamino)-1-(4-methoxyphenyl)ethyl] cyclohexanol hydrochloride.
Chemical name: 1-[(1RS)-2-(Dimethylamino)-1-(4-methoxyphenyl)ethyl] cyclohexanol hydrochloride.