

What is in this leaflet
This leaflet answers some common questions about ARCOXIA. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking ARCOXIA against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What ARCOXIA is used for
ARCOXIA is used for the following:
- to treat the symptoms of osteoarthritis
- to treat gout attacks
- to relieve short term pain, including cramp-like pain or discomfort before or during a menstrual period, and pain associated with minor dental procedures.
Osteoarthritis
Osteoarthritis is a joint disease. It results from the gradual breakdown of the cartilage that covers the joints and cushions the ends of bones.
Symptoms of osteoarthritis include pain, tenderness, stiffness of one or more joints, and physical disability. The hips and knees are the most commonly affected joints, but other joints, such as those of the hands and spine, may also be affected.
Osteoarthritis is more common in women than in men. Many factors can lead to the development of osteoarthritis, including obesity and joint injury (e.g. from sport).
Gout
Gout is a disease that causes attacks of arthritis, usually in a single joint. The affected joint is red, swollen, painful and extremely tender. A gout attack usually lasts a few days and it may recur.
For more information about osteoarthritis or gout, contact the Arthritis Foundation in the capital city of your state.
How ARCOXIA works
ARCOXIA belongs to a group of medicines called Coxibs. It works in a similar way to traditional anti-inflammatory medicines, known as Non-Steroidal Anti-Inflammatory Drugs (NSAIDs), by blocking the production of substances that cause pain and inflammation. In clinical trials, ARCOXIA has been shown to have a lower risk of serious side effects on the stomach (for example, bleeding stomach ulcers) than NSAIDs. However taking aspirin with ARCOXIA may reverse this benefit.
Your doctor may have prescribed ARCOXIA for another reason. Ask your doctor if you have any questions about why ARCOXIA has been prescribed for you.
The safety and effectiveness of ARCOXIA in children and teenagers under the age of 18 years have not been established.
ARCOXIA is not addictive.
Before you take ARCOXIA
When you must not take it
Do not take ARCOXIA if:
- you have an allergy to ARCOXIA or any of the ingredients listed at the end of this leaflet
- you have taken aspirin or other anti-inflammatory medicines (commonly known as NSAIDs) before, which caused asthma, pinkish itchy swellings on the skin (hives), runny nose, or other allergic reactions
- you have had heart failure, a heart attack, chest pain (angina), narrow or blocked arteries of the extremities (peripheral arterial disease), a stroke or mini stroke (TIA or transient ischaemic attack)
- you have had or are having major surgery on your heart or arteries
- you have high blood pressure that has not been controlled by treatment (check with your doctor or nurse if you are not sure whether your blood pressure is adequately controlled)
- you have serious liver disease
- you have a current stomach ulcer or bleeding in your stomach or intestines
- you have serious kidney disease
Do not take ARCOXIA if:
- the packaging is torn or shows signs of tampering
- the expiry date on the pack has passed
If you take this medicine after the expiry date has passed, it may not work.
If you are not sure whether you should start taking ARCOXIA, talk to your doctor.
Before you start to take it
Discuss the signs and/or symptoms of serious cardiovascular risks and steps to take if they occur with your doctor.
Tell your doctor if:
- you are pregnant or intend to become pregnant
ARCOXIA should not be used from 20 weeks of pregnancy onwards because it may harm the fetus. If there is a need to consider using ARCOXIA during your pregnancy, your doctor will discuss with you the benefits and risks of using it.
- you are breast-feeding or plan to breast-feed
It is not known if ARCOXIA passes into breast milk. You and your doctor should discuss whether you should stop breast-feeding or not take ARCOXIA.
- you have or have had any medical conditions, especially the following:
- kidney disease
- liver disease
- asthma, hives, itching, or skin rash
- dehydration, for example by a prolonged bout of vomiting or diarrhoea
- Inflammatory Bowel Disease
- heart failure
- high blood pressure
- heart attack, angina, or a blocked artery in your heart
- swelling of the ankles, feet or legs due to fluid retention (also called oedema)
- narrow or blocked arteries of the extremities
- stroke or mini stroke
- conditions which increase your risk of heart disease such as diabetes, high cholesterol, smoking or a first degree relative with heart disease
- you have a history of ulcers or bleeding in your stomach or intestines
- you have an infection
If you take ARCOXIA while you have an infection, it may hide fever and may make you think, mistakenly, that you are better or that your infection is less serious than it might be.
- you have any allergies to any other medicines or any other substances, such as foods, preservatives or dyes.
If you have not told your doctor about any of the above, tell them before you take any ARCOXIA.
Taking other medicines
Tell your doctor if you are taking any other medicines, including medicines that you buy without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and ARCOXIA may interfere with each other. These include:
- warfarin, used to prevent blood clots
- ACE inhibitor and angiotensin receptor blocker medicines used to lower high blood pressure or treat heart failure
- some fluid tablets (diuretics)
- lithium, used to treat mood swings and some types of depressions
- methotrexate, used to treat arthritis and some types of cancer
- rifampicin, an antibiotic used to treat tuberculosis and other infections
- ethinyl oestradiol, used in some oral contraceptives
- ketoconazole, used to treat fungal infections
- conjugated estrogens, used in some hormone replacement therapies
These medicines may be affected by ARCOXIA or may affect how well it works. You may need different amounts of your medicine, or you may need to take different medicines.
If you are taking low-dose aspirin to help prevent a heart attack or stroke, do not stop taking it unless your doctor tells you to because ARCOXIA cannot replace the use of aspirin for this purpose.
Some medicines should not be taken with ARCOXIA. These include:
- non-steroidal anti-inflammatory medicines (NSAIDs), used to relieve pain, swelling, and other symptoms of inflammation
- aspirin when used regularly for conditions other than to prevent heart attack or stroke.
Your doctor or pharmacist has more information on medicines to be careful with or avoid while taking ARCOXIA.
How to take ARCOXIA
How much to take
Take ARCOXIA only when prescribed by your doctor.
For osteoarthritis, the recommended dose is 30mg once a day, increased to a maximum of 60mg once a day if needed.
For the relief of gout attacks, the recommended dose is 120 mg taken once a day, which should only be used during the painful period, to a maximum of 8 days treatment.
For the relief of cramp-like pain or discomfort before or during a menstrual period, the recommended dose is 120 mg taken once a day, which should only be used during the painful period, to a maximum of 8 days treatment.
For the relief of short term pain after dental procedures, the recommended dose is 90 mg taken once a day, which should only be used during the painful period, to a maximum of 8 days treatment.
Doses greater than those recommended for each condition stated above have either not been shown to improve the effectiveness of ARCOXIA or have not been studied. Therefore, the daily doses stated above for each condition should not be exceeded.
If you have mild liver disease, do not take more than 60 mg once a day.
If you have moderate liver disease, do not take more than 60 mg every second day or 30mg once a day.
Swallow ARCOXIA with a glass of water.
Follow all directions given to you by your doctor carefully. They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
When to take it
Take your ARCOXIA at about the same time each day. Taking ARCOXIA at the same time each day will have the best effect. It will also help you remember when to take the dose.
It does not matter if you take ARCOXIA before, with or after food.
How long to take it
Depending on your condition, you may need to take ARCOXIA for a few days or for a longer period.
For arthritis ARCOXIA helps relieve your symptoms but it does not cure it. Continue taking ARCOXIA for as long as your doctor prescribes it.
For the relief of gout attacks and short term pain, including menstrual pain and dental pain, ARCOXIA should only be used during the painful period, limited to a maximum of 8 days treatment.
If you are not sure how long to take ARCOXIA, talk to your doctor.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to. Otherwise, take it as soon as you remember, and then go back to taking your tablet(s) as you would normally.
If you are not sure whether to skip the dose, talk to your doctor or pharmacist.
Do not take a double dose to make up for the dose that you missed.
If you have trouble remembering to take your tablets, ask your pharmacist for some hints.
If you take too much (overdose)
Immediately telephone your doctor or Poisons Information Centre (in Australia, telephone 131 126; in New Zealand, telephone 0800 POISON or 0800 764 766), or go to accident and emergency at your nearest hospital, if you think that you or anyone else may have taken too much ARCOXIA. Do this even if there are no signs of discomfort or poisoning.
While you are using ARCOXIA
Things you must do
If you become pregnant while taking ARCOXIA, tell your doctor immediately.
If you get an infection while taking ARCOXIA, tell your doctor. ARCOXIA may hide fever and may make you think, mistakenly, that you are better or that that your infection is less serious than it might be.
If you notice any of the following, tell your doctor immediately:
- vomiting blood or material that looks like coffee grounds, bleeding from the back passage, black sticky bowel motions (stools), or bloody diarrhoea
These symptoms may occur at any time during use and without warning.
- any symptoms that could indicate a severe allergic reaction such as inability to breath or a serious skin reaction which may occur without warning
You may need urgent medical attention.
If any of the following symptoms: serious skin rash, shortness of breath, chest pains or ankle swelling, appear or worsen, stop your treatment with ARCOXIA and consult a doctor, as soon as is practical.
If you are about to be started on any new medicine, tell your doctor and pharmacist that you are taking ARCOXIA.
Additional things to be aware of while taking ARCOXIA:
ARCOXIA works equally well in older and younger adult patients. Adverse experiences may occur at a higher incidence in older patients compared to younger patients. If you are elderly (i.e., over 65 years of age), your doctor will want to keep a regular check on you. No dosage adjustment is necessary for older patients.
If you have kidney, liver or heart disease, your doctor will want to keep a regular check on you.
Your doctor will want to discuss your treatment from time to time. It is important that you use the lowest dose that controls your pain and you should not take ARCOXIA for longer than necessary. This is because the risk of heart attacks and strokes might increase after prolonged treatment, especially with high doses.
ARCOXIA can increase blood pressure in some people, especially in high doses, and this could increase the risk of heart attacks and strokes. Your doctor will want to check your blood pressure from time to time, to make sure that it is safe to continue treatment.
Things you must not do
Do not give ARCOXIA to anyone else, even if they have the same condition as you.
Things to be careful of
Be careful driving or operating machinery until you know how ARCOXIA affects you. The effect of ARCOXIA on the ability to drive a car or operate machinery has not been studied, although it is thought to be unlikely to have any effect on these activities. However, as with many medicines, ARCOXIA may cause certain side effects in some people, including dizziness and tiredness. Make sure you know how you react to ARCOXIA before you drive a car or operate machinery.
Things that would be helpful for your arthritis
Some self help measures suggested below may help your condition. Talk to your doctor or pharmacist about these measures and for more information.
- Exercise
Regular exercise can help reduce pain and disability from arthritis by increasing muscle strength and reducing the load on joints, but it is important not to overdo it. Walking is good exercise, however, before starting any exercise, ask your doctor about the best kind of programme for you. - Weight
Your doctor may suggest losing some weight to help reduce the strain on your joints. Some people may need a dietician's help to lose weight. - Hot and cold treatments
These can help to relieve pain from arthritis, including hot and cold packs, or exercising in a warm water pool (hydrotherapy). Talk to your doctor or physiotherapist about the types of hot and cold treatments available that would be helpful for your condition. - Support devices and aids
These may assist in coping with everyday tasks at home or help to relieve pain in certain joints. Ask a physiotherapist or occupational therapist for advice about joint protection, aids and special equipment.
Side Effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking ARCOXIA.
ARCOXIA helps most people with arthritis, menstrual pain, or other types of pain, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice or have any of the following and they worry you:
- feeling sick (nausea), vomiting
- heartburn, uncomfortable feeling in the stomach
- mouth ulcers
- changes in taste
- diarrhoea
- swelling of the legs, ankles or feet
- high/increase in blood pressure
- headache, dizziness
- unusual tiredness or weakness
- difficulty sleeping
- depression
- restlessness
- signs of urinary tract infection, including painful burning when passing urine
- high levels of potassium in your blood
- signs of an infection of the breathing passages, including runny nose, sore throat, cough
- feeling anxious
- confusion
- seeing, feeling or hearing things that are not there
- blurred vision
These are the more common side effects of ARCOXIA.
Tell your doctor immediately if you notice any of the following:
- skin rash or itchiness
- pinkish, itchy swellings on the skin, also called hives or nettlerash
- redness of the skin
- severe stomach pain
- passing little or no urine
- yellowing of the skin and/or eyes, also called jaundice
- a feeling of pain, tightness, pressure or heaviness in the chest (angina)
- increased tendency to bleed/bruise, or tendency to bleed for a longer period of time – these may be symptoms of reduced platelets in your body
- severe pain and swelling following a dental extraction, "dry socket"
- gout pain not relieved by ARCOXIA
These may be serious side effects. Some of these may be symptoms of an allergic reaction to ARCOXIA. You may need urgent medical attention.
If any of the following happen, tell your doctor immediately or go to accident and emergency at your nearest hospital:
- fever, skin rash, swelling of the face
- not enough amniotic fluid in the womb (diagnosed by a health care provider)
- kidney injury in newborns (diagnosed by a health care provider)
- swelling of the face, lips, mouth, throat or tongue which may cause difficulty in breathing or swallowing
- vomiting blood or material that looks like coffee grounds, bleeding from the back passage, black sticky bowel motions (stools), or bloody diarrhoea - these may be symptoms of bleeding stomach ulcers which may occur at any time during use and without warning
- shortness of breath, wheezing, or trouble breathing
- fast or irregular heartbeats, also called palpitations
- rare skin condition with severe blisters and bleeding in the lips, eyes, mouth, nose and genitals
- liver disease including liver failure with symptoms such as loss of appetite, yellowing of the skin and eyes, and dark coloured urine
- blood clots in the blood vessels of the lung and/or leg, and bleeding within the brain.
These may be serious side effects. You may need urgent medical attention.
Other side effects not listed above may also occur in some patients. Tell your doctor if you notice any other effects.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
After using ARCOXIA
Storage
Keep your tablets in the blister pack until it is time to take them. If you take the tablets out of the blister pack, they may not keep well.
Keep ARCOXIA in a cool dry place where the temperature stays below 30°C. Do not store it or any other medicine in the bathroom or near a sink.
Do not leave it in the car or on window sills. Heat and dampness can destroy some medicines.
Keep it where children cannot reach it. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking ARCOXIA or the tablets have passed their expiry date, ask your pharmacist what to do with any that are left over.
Product description
What it looks like
ARCOXIA comes as four strengths of tablets:
- 30 mg tablet - blue-green, apple-shaped biconvex film coated tablet with 101 marked on one side and ACX 30 on the other. Available in packs of 5 tablets*, 10 tablets (starter pack)*, and 30 tablets.
- 60 mg tablet - dark green, apple-shaped biconvex film coated tablet with 200 marked on one side and ARCOXIA 60 on the other. Available in packs of 5 tablets*, 10 tablets*, and 30 tablets.
- 90 mg tablet - white, apple-shaped biconvex film coated tablet debossed 202 marked on one side and ARCOXIA 90 on the other. Available in packs of 2 tablets (starter pack)*, 5 tablets*, and 10 tablets*.
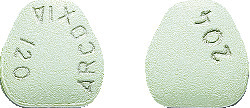
- 120 mg tablet - pale green, apple-shaped biconvex film coated tablet with 204 marked on one side and ARCOXIA 120 on the other. Available in packs of 2 tablets (starter pack)*, 5 tablets*, and 10 tablets.
*Presentation not currently marketed in Australia.
Ingredients
Active ingredient:
- ARCOXIA 30 mg tablet contains 30 mg etoricoxib
- ARCOXIA 60mg tablet contains 60 mg etoricoxib
- ARCOXIA 90 mg tablet contains 90 mg etoricoxib
- ARCOXIA 120 mg tablet contains 120 mg etoricoxib
Inactive ingredients:
- calcium hydrogen phosphate
- microcrystalline cellulose
- lactose monohydrate
- croscarmellose sodium
- hypromellose
- magnesium stearate
- carnauba wax
- titanium dioxide
- triacetin
- iron oxide yellow CI77492 (30mg, 60mg and 120mg tablets)
- indigo carmine CI73015 (30mg, 60mg and 120mg tablets)
ARCOXIA does not contain gluten, sucrose, tartrazine or any other azo dyes.
Manufacturer/Supplier
ARCOXIA is marketed in Australia by:
Organon Pharma Pty Ltd
Building A
26 Talavera Road
Macquarie Park, NSW 2113
Date of Preparation
This leaflet was last revised in May 2023.
Australian Register Numbers:
30mg tablet - AUST R 131797
60mg tablet - AUST R 81456
90mg tablet - AUST R 204147
120mg tablet - AUST R 81458
This CMI leaflet was current at the time of printing. To check if it has been updated, please view our website https://organon.com/australia or ask your pharmacist.
RCN100000101 - AU
S-WPPI-OG0663-T-012023
Published by MIMS June 2023
 The following lists additional adverse experiences, regardless of causality, occurring in patients receiving Arcoxia with a frequency of between 0.1% and less than 2.0% and at least 0.1% more frequently than in the placebo treatment group in ten placebo controlled studies of 6 to 12 weeks duration conducted in patients with OA at the therapeutically recommended doses (30 mg and 60 mg).
The following lists additional adverse experiences, regardless of causality, occurring in patients receiving Arcoxia with a frequency of between 0.1% and less than 2.0% and at least 0.1% more frequently than in the placebo treatment group in ten placebo controlled studies of 6 to 12 weeks duration conducted in patients with OA at the therapeutically recommended doses (30 mg and 60 mg). The EDGE and EDGE II studies compared the GI tolerability of etoricoxib 90 mg daily (1.5 to 3 times the doses recommended for OA) and diclofenac 150 mg daily in 7,111 patients with OA (EDGE study; mean duration of treatment 9 months) and 4,086 patients with RA (EDGE II; mean duration of treatment 19 months). In each of these studies, the adverse experience profile on Arcoxia was generally similar to that reported in the phase IIb/III placebo controlled clinical studies; however, hypertension and oedema related adverse events occurred at a higher rate on etoricoxib than on diclofenac. The rate of confirmed thrombotic cardiovascular serious adverse events occurring in the two treatment groups was similar.
The EDGE and EDGE II studies compared the GI tolerability of etoricoxib 90 mg daily (1.5 to 3 times the doses recommended for OA) and diclofenac 150 mg daily in 7,111 patients with OA (EDGE study; mean duration of treatment 9 months) and 4,086 patients with RA (EDGE II; mean duration of treatment 19 months). In each of these studies, the adverse experience profile on Arcoxia was generally similar to that reported in the phase IIb/III placebo controlled clinical studies; however, hypertension and oedema related adverse events occurred at a higher rate on etoricoxib than on diclofenac. The rate of confirmed thrombotic cardiovascular serious adverse events occurring in the two treatment groups was similar. The dose ranging study, Protocol 007, included 617 OA patients randomised to placebo or etoricoxib doses of 5, 10, 30, 60 and 90 mg. Protocol 007 demonstrated that all etoricoxib doses (5, 10, 30, 60, 90 mg) were significantly better than placebo for the 3 primary endpoints: the WOMAC pain subscale, the Investigator Global Assessment of Disease Status, and the Patient Global Assessment of Response to Therapy, as shown in the accompanying figure (see Figure 1). The 60 mg dose met three out of three predefined efficacy criteria, whereas the 30 mg dose met two out of three predefined efficacy criteria; and etoricoxib 60 mg demonstrated significantly greater efficacy than etoricoxib 30 mg. Etoricoxib 90 mg did not demonstrate greater efficacy than the etoricoxib 60 mg dose.
The dose ranging study, Protocol 007, included 617 OA patients randomised to placebo or etoricoxib doses of 5, 10, 30, 60 and 90 mg. Protocol 007 demonstrated that all etoricoxib doses (5, 10, 30, 60, 90 mg) were significantly better than placebo for the 3 primary endpoints: the WOMAC pain subscale, the Investigator Global Assessment of Disease Status, and the Patient Global Assessment of Response to Therapy, as shown in the accompanying figure (see Figure 1). The 60 mg dose met three out of three predefined efficacy criteria, whereas the 30 mg dose met two out of three predefined efficacy criteria; and etoricoxib 60 mg demonstrated significantly greater efficacy than etoricoxib 30 mg. Etoricoxib 90 mg did not demonstrate greater efficacy than the etoricoxib 60 mg dose. The effectiveness of Arcoxia 60 mg was shown to be comparable to naproxen 500 mg twice daily for treatment of the signs and symptoms of OA (see Table 4). Onset of efficacy was evaluated only in the 60 mg studies and was demonstrated within the first 24 hours of initiating treatment. In patients with OA of the hand, reductions in pain and stiffness, and improvement in physical function, as measured by the AUSCAN questionnaire were superior to placebo and similar to that in patients treated with naproxen.
The effectiveness of Arcoxia 60 mg was shown to be comparable to naproxen 500 mg twice daily for treatment of the signs and symptoms of OA (see Table 4). Onset of efficacy was evaluated only in the 60 mg studies and was demonstrated within the first 24 hours of initiating treatment. In patients with OA of the hand, reductions in pain and stiffness, and improvement in physical function, as measured by the AUSCAN questionnaire were superior to placebo and similar to that in patients treated with naproxen. The clinical benefit of both Arcoxia 30 mg and 60 mg was maintained for the duration of the studies.
The clinical benefit of both Arcoxia 30 mg and 60 mg was maintained for the duration of the studies.
 GI tolerability, defined as patients discontinuing the study for any clinical (e.g. dyspepsia, abdominal pain, ulcer) or laboratory (e.g. increased ALT, AST) GI adverse experience including hepatic events, was also evaluated in each individual study within the MEDAL program. The EDGE and EDGE II studies assessed GI tolerability as the primary endpoint. They compared etoricoxib 90 mg daily and diclofenac 150 mg daily in patients with OA (EDGE) and RA (EDGE II). The MEDAL study compared GI tolerability between etoricoxib 60 mg (OA) or 90 mg (OA and RA) to diclofenac 150 mg daily as a secondary objective. In all three studies, etoricoxib demonstrated superior GI tolerability compared to diclofenac (p-values < 0.001; see Figure 2). The GI tolerability benefit for etoricoxib was significant both for the clinical and for the laboratory components that make up this composite endpoint.
GI tolerability, defined as patients discontinuing the study for any clinical (e.g. dyspepsia, abdominal pain, ulcer) or laboratory (e.g. increased ALT, AST) GI adverse experience including hepatic events, was also evaluated in each individual study within the MEDAL program. The EDGE and EDGE II studies assessed GI tolerability as the primary endpoint. They compared etoricoxib 90 mg daily and diclofenac 150 mg daily in patients with OA (EDGE) and RA (EDGE II). The MEDAL study compared GI tolerability between etoricoxib 60 mg (OA) or 90 mg (OA and RA) to diclofenac 150 mg daily as a secondary objective. In all three studies, etoricoxib demonstrated superior GI tolerability compared to diclofenac (p-values < 0.001; see Figure 2). The GI tolerability benefit for etoricoxib was significant both for the clinical and for the laboratory components that make up this composite endpoint. Hepatic related adverse events resulting in discontinuation were evaluated in each individual study within the MEDAL program. The incidences of discontinuations were significantly lower in the etoricoxib 60 and 90 mg treatment groups compared with the diclofenac 150 mg treatment groups for both OA and RA patients in all three studies.
Hepatic related adverse events resulting in discontinuation were evaluated in each individual study within the MEDAL program. The incidences of discontinuations were significantly lower in the etoricoxib 60 and 90 mg treatment groups compared with the diclofenac 150 mg treatment groups for both OA and RA patients in all three studies.
 Both endoscopy studies included the following patients at a higher risk for GI ulcers: patients with active Helicobacter pylori infection; baseline gastroduodenal erosions; prior history of perforation, ulcer or bleed (PUB); and/or concomitant use of corticosteroids. Four hundred patients (28%) were 65 years of age and older. The advantage of Arcoxia versus naproxen or ibuprofen was maintained in these higher risk subgroups.
Both endoscopy studies included the following patients at a higher risk for GI ulcers: patients with active Helicobacter pylori infection; baseline gastroduodenal erosions; prior history of perforation, ulcer or bleed (PUB); and/or concomitant use of corticosteroids. Four hundred patients (28%) were 65 years of age and older. The advantage of Arcoxia versus naproxen or ibuprofen was maintained in these higher risk subgroups.
