1 Name of Medicine
Aripiprazole (as monohydrate).
2 Qualitative and Quantitative Composition
Aripiprazole is present in Aripena as aripiprazole (as monohydrate). Aripena is presented as a therapeutic kit. The presentation contains a sterile, single-dose, lyophilised powder for reconstitution with water for injections to give a prolonged-release suspension for injection to deliver 400 mg of aripiprazole per vial strength.
3 Pharmaceutical Form
Aripena (aripiprazole) 400 mg powder and diluent for prolonged-release suspension for injection (therapeutic kit).
4.1 Therapeutic Indications
For the acute and maintenance treatment of schizophrenia in adults.
For maintenance treatment to prevent the recurrence of manic or mixed episodes of bipolar I disorder in adult patients as monotherapy.
4.2 Dose and Method of Administration
Recommended dosage and dosage adjustment.
Aripena is intended for intramuscular injection only. For patients who have never taken aripiprazole, establish tolerability with oral aripiprazole prior to initiating treatment with Aripena. Due to the half-life of oral aripiprazole, it may take up to 2 weeks to fully assess tolerability. The recommended starting and maintenance dose of Aripena is 400 mg. Titration of the dose of Aripena is not required. Aripena is to be administered by a healthcare professional only, once-monthly, as a single injection into the deltoid or gluteal muscle (no sooner than 26 days after the previous injection). Sites of injections should be rotated between the two gluteal or deltoid muscles. After the first Aripena injection, treatment with oral aripiprazole (10 mg to 20 mg), or other oral antipsychotic, should be continued for 14 consecutive days to maintain therapeutic antipsychotic concentrations during initiation of therapy.
If there are adverse reactions with the 400 mg dose, reduction of the dose to *300 mg once-monthly should be considered.
*300 mg is available in another brand.
Switching from oral antipsychotics.
For patients who have never taken oral or injectable aripiprazole, establish tolerability with oral aripiprazole prior to initiating treatment with Aripena. When switching from oral antipsychotics, patients may continue their current oral antipsychotic (oral aripiprazole or prescribed dose of other oral antipsychotic) for 14 days following the first dose of Aripena to maintain therapeutic plasma concentrations during the initiation of Aripena. Aripena should then be administered once monthly as described above.
Switching from long-acting injectable antipsychotics.
For patients who have never taken oral or injectable aripiprazole, establish tolerability with oral aripiprazole prior to initiating treatment with Aripena. When switching patients from previous long-acting injectable antipsychotics, initiate Aripena therapy in place of the next scheduled injection, with 14 days of concurrent oral aripiprazole. Aripena should then be continued monthly.
Discontinuation of Aripena.
If Aripena is discontinued, its prolonged-release characteristics must be considered.
Missed doses.
See Table 1.

Dosage in special populations.
Elderly population.
The effectiveness and safety of Aripena in the treatment of schizophrenia in patients ≥ 61 years and in the treatment of bipolar I disorder in patients ≥ 66 years have not been evaluated.
Renal impairment.
No dosage adjustment of Aripena is required for patients with renal impairment. See Section 5.2 Pharmacokinetic Properties, Special populations.
Hepatic impairment.
Based on oral data no dosage adjustment of Aripena is required for patients with mild or moderate hepatic impairment. In patients with severe hepatic impairment, the data available are insufficient to establish dosage recommendations. In these patients dosing should be managed cautiously; use of oral aripiprazole should be considered. See Section 5.2 Pharmacokinetic Properties, Special populations.
Other special populations.
No dosage adjustment of Aripena is recommended based on gender, race or smoking status.
Known CYP2D6 poor metabolisers.
In patients who are known to be CYP2D6 poor metabolisers, the starting and maintenance dose of Aripena should be 300 mg. If Aripena is taken concomitantly with strong CYP3A4 inhibitors, the dose of Aripena should be reduced to 200 mg. See Section 4.5 Interactions with Other Medicines and Other Forms of Interactions.
Paediatric population.
The safety and efficacy of Aripena in children and adolescents aged 0-17 years have not been established. No data are available.
Dose adjustments due to interactions.
Dosage adjustments are recommended in patients taking concomitant strong CYP3A4 inhibitors or strong CYP2D6 inhibitors for more than 14 days. If the CYP3A4 inhibitor or CYP2D6 inhibitor is withdrawn, the Aripena dose may need to be increased. See Table 2. Avoid concomitant use of CYP3A4 inducers with Aripena for more than 14 days, because the blood levels of aripiprazole will be decreased and may fall below the effective levels. See Table 2.

Administration.
Aripena is presented as a therapeutic kit. See instructions below for reconstitution and injection.
Therapeutic kit. Aripena is available in a therapeutic kit which contains one vial of lyophilised powder, one vial of diluent (water for injections), one 3 mL sterile syringe with a 21 gauge needle for reconstitution, one sterile syringe without a needle, one 1-inch (25 mm) 23 gauge sterile safety needle for injection, one 1.5-inch (38 mm) 22 gauge sterile safety needle for injection, one 2-inch (51 mm) 21 gauge sterile safety needle for injection and one vial adapter.
For deep intramuscular deltoid or gluteal injection only: do not administer intravenously or subcutaneously.
The suspension should be injected immediately after reconstitution, but can be stored below 25°C for up to 4 hours in the vial. If the injection is not performed immediately after reconstitution, shake the vial vigorously for at least 60 seconds to re-suspend prior to the injection. Aripena should be administered by a healthcare professional only, once-monthly. The injection should be injected slowly as a single injection (doses must not be divided) into the deltoid or gluteal muscle.
Any unused product or waste material should be disposed of in accordance with local requirements.
Step 1: preparation prior to reconstitution of the lyophilised Aripena powder.
a) Lay out the contents of the package and confirm that all components listed below are provided:
One vial containing aripiprazole 400 mg (as monohydrate) powder for injection;
One vial containing water for injections 2.0 mL;
One 3 mL luer lock syringe with pre-attached 21 gauge, 1.5-inch (38 mm) hypodermic safety needle with needle protection device;
One 3 mL disposable syringe with luer lock tip;
One vial adapter;
One 23 gauge, 1-inch (25 mm) hypodermic safety needle with needle protection device;
One 22 gauge, 1.5-inch (38 mm) hypodermic safety needle with needle protection device; and
One 21 gauge, 2-inch (51 mm) hypodermic safety needle with needle protection device.
b) The contents of the vial containing aripiprazole (as monohydrate) lyophilised powder should be suspended using the water for injections supplied in the carton.
c) The vials containing aripiprazole (as monohydrate) lyophilised powder and water for injections are for single-use only.
d) Use appropriate aseptic techniques throughout the reconstitution and reconstitute at room temperature.
e) Select the amount of water for injections needed for reconstitution at room temperature. See Table 3.
 It is important to note that there is more water for injections in the vial than is needed to reconstitute the contents of the vial containing aripiprazole (as monohydrate) lyophilised powder for prolonged-release suspension for injection.
It is important to note that there is more water for injections in the vial than is needed to reconstitute the contents of the vial containing aripiprazole (as monohydrate) lyophilised powder for prolonged-release suspension for injection.
Step 2: reconstitution of the lyophilised powder.
a) Remove the cap of the vial of water for injections and remove the cap of the vial containing the aripiprazole (as monohydrate) lyophilised powder and wipe the tops with a sterile alcohol swab.
b) Using the syringe with pre-attached hypodermic safety needle, withdraw the pre-determined water for injections volume from the vial of water for injections into the syringe. A small amount of residual water for injections will remain in the vial following withdrawal.
c) Slowly inject the water for injections into the vial containing the aripiprazole (as monohydrate) lyophilised powder.
d) Withdraw air to equalise the pressure in the vial by pulling back slightly on the plunger. Subsequently, remove the needle from the vial. Engage the needle safety device by using the one-handed technique. Gently press the sheath against a flat surface until the needle is firmly engaged in the needle protection sheath. Visually confirm that the needle is fully engaged into the needle protection sheath, and discard. Shake the vial vigorously for 30 seconds until the suspension appears uniform.
e) Visually inspect the reconstituted suspension for particulate matter and discolouration prior to administration. The reconstituted Aripena is a uniform, homogeneous suspension that is opaque and milky-white in colour. Do not use if reconstituted suspension contains particulate matter or any discolouration.
f) If the injection is not performed immediately after reconstitution, keep the vial below 25°C for up to 4 hours and shake the vial vigorously for at least 60 seconds to re-suspend prior to injection.
g) Do not store the reconstituted suspension in the syringe.
Step 3: preparation prior to injection.
a) Use appropriate aseptic techniques throughout injection of the reconstituted Aripena suspension.
b) Remove the cover from the vial adapter package. Do not remove the vial adapter from the package.
c) Using the vial adapter package to handle the vial adapter, attach the pre-packaged disposable syringe to the vial adapter.
d) Use the disposable syringe to remove the vial adapter from the package and discard the vial adapter package. Do not touch the spike tip of the adapter at any time.
e) Determine the recommended volume for injection. See Table 4.
 f) Wipe the top of the vial of the reconstituted Aripena suspension with a sterile alcohol swab.
f) Wipe the top of the vial of the reconstituted Aripena suspension with a sterile alcohol swab.
g) Place and hold the vial of the reconstituted Aripena suspension on a hard surface. Attach the adapter-syringe assembly to the vial by holding the outside of the adapter and pushing the adapter's spike firmly through the rubber stopper, until the adapter snaps in place.
h) Slowly withdraw the recommended volume from the vial into the disposable syringe to allow for injection. A small amount of excess product will remain in the vial.
Step 4: injection procedure.
a) Detach the disposable syringe containing the recommended volume of reconstituted Aripena suspension from the vial.
b) Select one of the hypodermic safety needles listed in Table 5 according to the injection site and patient's weight.
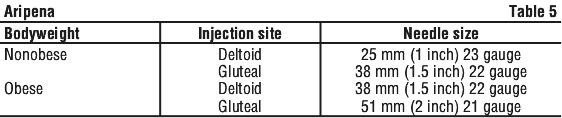 Attach the needle to the disposable syringe containing the suspension for injection. To avoid subcutaneous drug administration, examine the depth of subcutaneous fat at the injection site and select the appropriate needle size. Ensure the needle is firmly seated on the needle safety device with a push and clockwise twist and then pull the needle cap straight away from the needle.
Attach the needle to the disposable syringe containing the suspension for injection. To avoid subcutaneous drug administration, examine the depth of subcutaneous fat at the injection site and select the appropriate needle size. Ensure the needle is firmly seated on the needle safety device with a push and clockwise twist and then pull the needle cap straight away from the needle.
Slowly inject the recommended volume as a single intramuscular injection into the deltoid or gluteal muscle. Do not massage the injection site. Do not administer intravenously or subcutaneously.
Step 5: procedures after injection.
a) Engage the needle safety device as described in Step 2 (d). Dispose of the vials, adapter, needles, and syringe appropriately after injection. The vials containing aripiprazole (as monohydrate) lyophilised powder and water for injections are for single-use only.
b) Rotate sites of injections between the two deltoid or gluteal muscles. Look for signs or symptoms of inadvertent intravenous administration.4.3 Contraindications
Hypersensitivity to aripiprazole or any of the excipients. See Section 2 Qualitative and Quantitative Composition.
4.4 Special Warnings and Precautions for Use
Elderly patients with dementia-related psychosis.
Increased mortality.
Elderly patients with dementia-related psychosis treated with atypical antipsychotic drugs, including aripiprazole, are at an increased risk of death compared to placebo. Analyses of 17 placebo-controlled trials (modal duration of 10 weeks) in these patients revealed a risk of death in the drug-treated patients of between 1.6 to 1.7 times that seen in placebo-treated patients. Over the course of a typical 10-week controlled trial, the rate of death in drug-treated patients was about 4.5%, compared to a rate of about 2.6% in the placebo group. Although the causes of death varied, most of the deaths appeared to be either cardiovascular (e.g. heart failure, sudden death) or infectious (e.g. pneumonia) in nature.
In three placebo-controlled trials with oral aripiprazole in elderly patients with psychosis associated with Alzheimer's disease (n=938; mean age: 82.4 years; range: 56-99 years), patients treated with aripiprazole were at an increased risk of death compared to placebo. The rate of death in the aripiprazole-treated patients was 3.5% compared with 1.7% in the placebo group. Aripiprazole is not indicated for the treatment of patients with dementia-related psychosis.
Cerebrovascular adverse events.
In the same three 10-week placebo-controlled trials, cerebrovascular adverse reactions (e.g. stroke, transient ischaemic attack), including fatalities, were reported in patients (mean age: 84 years; range: 78-88 years). Overall, 1.3% of aripiprazole-treated patients reported cerebrovascular adverse reactions compared with 0.6% of placebo-treated patients in these trials. However, in one of these trials, a fixed-dose trial, there was a significant dose response relationship for cerebrovascular adverse reactions in patients treated with aripiprazole. The safety and efficacy of aripiprazole on the treatment of patients with psychosis associated with dementia have not been established. Aripiprazole is not indicated for the treatment of patients with dementia-related psychosis.
General.
During antipsychotic treatment, improvement in the patient's clinical condition may take several days to some weeks. Patients should be closely monitored during this period.
Suicide.
The possibility of a suicide attempt is inherent in psychotic illnesses and close supervision of high-risk patients should accompany drug therapy. As aripiprazole is to be administered by a healthcare professional, suicide due to an overdose is considered unlikely.
Tardive dyskinesia.
In clinical trials of one year or less duration, there were uncommon reports of treatment-emergent tardive dyskinesia during treatment with aripiprazole. The risk of tardive dyskinesia increases with long-term exposure to antipsychotic treatment. If signs and symptoms of tardive dyskinesia appear in a patient on aripiprazole, dose reduction or discontinuation of treatment should be considered (see Section 4.2 Dose and Method of Administration). These symptoms can temporally deteriorate or can even arise after discontinuation of treatment.
Neuroleptic malignant syndrome.
Neuroleptic malignant syndrome (NMS) is a potentially fatal symptom complex associated with antipsychotic medicinal products. In clinical trials, rare cases of NMS were reported during treatment with aripiprazole. Clinical manifestations of NMS are hyperpyrexia, muscle rigidity, altered mental status and evidence of autonomic instability (irregular pulse or blood pressure, tachycardia, diaphoresis and cardiac dysrhythmia). Additional signs may include elevated creatine phosphokinase, myoglobinuria (rhabdomyolysis), and acute renal failure. However, elevated creatine phosphokinase and rhabdomyolysis, not necessarily in association with NMS, have also been reported. If a patient develops signs and symptoms indicative of NMS, or presents with unexplained high fever without additional clinical manifestations of NMS, all antipsychotic medicinal products, including aripiprazole, must be discontinued.
Seizure.
In clinical trials, uncommon cases of seizures were experienced during treatment with aripiprazole. As with other antipsychotic drugs, aripiprazole should be used cautiously in patients who have a history of seizure disorder or have conditions associated with seizures.
Hyperglycaemia and diabetes mellitus.
Hyperglycaemia, in some cases extreme and associated with ketoacidosis or hyperosmolar coma or death, has been reported in patients treated with atypical antipsychotics including aripiprazole. In clinical trials, the observed differences in the incidence rates of hyperglycaemia-related adverse reactions (including diabetes) or in abnormal glycaemia laboratory values between aripiprazole (< 1%) and placebo (0%) could be considered as of no major clinical concern.
Assessment of the relationship between atypical antipsychotic use and glucose abnormalities is complicated by the possibility of an increased background risk of diabetes mellitus in patients with schizophrenia and the increasing incidence of diabetes mellitus in the general population. Given these confounders, the relationship between atypical antipsychotic use and hyperglycaemia-related adverse events is not completely understood. However, epidemiological studies suggest an increased risk of treatment-emergent hyperglycaemia-related adverse events in patients treated with atypical antipsychotics. Precise risk estimates for hyperglycaemia-related adverse events in patients treated with atypical antipsychotics are not available.
Patients with an established diagnosis of diabetes mellitus who are started on atypical antipsychotics, including aripiprazole, should be monitored regularly for worsening of glucose control. All patients who are starting treatment with atypical antipsychotics, including aripiprazole, should undergo fasting blood glucose testing at the beginning of treatment and periodically during treatment. Any patient treated with atypical antipsychotics, including aripiprazole, should be monitored for symptoms of hyperglycaemia including polydipsia, polyuria, polyphagia and weakness. Patients who develop symptoms of hyperglycaemia during treatment with atypical antipsychotics should undergo fasting blood glucose testing. In some cases, hyperglycaemia has resolved when the atypical antipsychotic was discontinued; however, some patients required continuation of anti-diabetic treatment despite discontinuation of the suspect drug.
In patients with significant treatment-emergent hyperglycaemia, discontinuation of aripiprazole should be considered.
Cardiovascular disorders.
Aripiprazole should be used with caution in patients with known cardiovascular disease (e.g. history of myocardial infarction or ischaemic heart disease, heart failure, or conduction abnormalities), cerebrovascular disease, or conditions which would predispose patients to hypotension (e.g. dehydration, hypovolaemia, and treatment with antihypertensive medications) or hypertension, including accelerated or malignant. Patients with a history of clinically significant cardiovascular disorders were excluded from clinical trials.
QT interval.
In clinical trials of treatment with aripiprazole IM depot and oral aripiprazole, the incidence of QT prolongation was uncommon. As with other antipsychotics, aripiprazole should be used with caution in patients with a family history of QT prolongation. Also see Section 4.8 Adverse Effects (Undesirable Effects), Laboratory tests, QT interval.
Orthostatic hypotension.
Aripiprazole may be associated with orthostatic hypotension, potentially due to its α1-adrenergic receptor antagonism. Aripiprazole may induce orthostatic hypotension, tachycardia, dizziness and sometimes syncope, especially at the initiation of treatment. In the double-blind controlled phase of the long-term clinical trials, orthostasis-related events were reported in 2/534 (0.4%) patients treated with aripiprazole. In the 12-week clinical trial in acutely relapsed patients, orthostasis-related events were reported in 1/167 (0.6%) patient treated with aripiprazole, while syncope and orthostatic hypotension each occurred and in 2/172 (1.2%) patients treated with placebo.
Venous thromboembolism.
Cases of venous thromboembolism (VTE) have been reported with antipsychotic drugs. Since patients treated with antipsychotics often present with acquired risk factors for VTE, all possible risk factors for VTE should be identified before and during treatment with aripiprazole and preventive measures undertaken.
Falls.
Somnolence, postural hypotension, motor and sensory instability have been reported with the use of antipsychotics, including aripiprazole, which may lead to falls. Caution should be taken when treating patients with diseases, conditions, or who are taking medications that could exacerbate these effects.
Body temperature regulation.
Disruption of the body's ability to increase or reduce core body temperature has been attributed to antipsychotic agents, including aripiprazole. Appropriate care is advised when prescribing aripiprazole for patients who will be experiencing conditions which may contribute to an elevation in core body temperature, e.g. exercising strenuously, exposure to extreme heat, receiving concomitant medication with anticholinergic activity or being subject to dehydration.
Patients should be advised regarding appropriate care in avoiding overheating and dehydration.
Dysphagia.
Oesophageal dysmotility and aspiration have been associated with antipsychotic drug use, including aripiprazole. Aripiprazole and other antipsychotic drugs should be used cautiously in patients at risk of aspiration pneumonia (e.g. elderly patients).
Akathisia.
Class effect.
The presentation of akathisia may be variable and comprises subjective complaints of restlessness and an overwhelming urge to move and either distress or motor phenomena (such as pacing, swinging of the legs while seated, rocking from foot to foot), or both. Particular attention should be paid to the monitoring for such symptoms and signs as, left untreated, akathisia is associated with poor compliance and an increased risk of relapse.
Leukopenia, neutropenia and agranulocytosis.
Class effect.
In clinical trial and/or post-marketing experience, events of leukopenia/neutropenia have been reported temporally related to antipsychotic agents, including aripiprazole. Agranulocytosis has also been reported.
Possible risk factors for leukopenia/neutropenia include pre-existing low white blood cell count (WBC) and history of drug-induced leukopenia/neutropenia. Patients with a history of a clinically significant low WBC or drug-induced leukopenia/neutropenia should have their complete blood cell (CBC) monitored frequently during the first few months of therapy, and discontinuation of aripiprazole should be considered at the first sign of a clinically significant decline in WBC in the absence of other causative factors.
Patients with clinically significant neutropenia should be carefully monitored for fever or other symptoms or signs of infection and treated promptly, if such symptoms or signs occur. Patients with severe neutropenia (absolute neutrophil count < 1000/mm3) should discontinue aripiprazole and have their WBC followed until recovery. See Section 4.8 Adverse Effects (Undesirable Effects), Laboratory tests.
Pathological gambling and impulse-control disorders.
Patients can experience increased urges, particularly for gambling, and the inability to control these urges while taking aripiprazole. Other urges reported include: increased sexual urges, compulsive spending, binge or compulsive eating, and other impulsive and compulsive behaviours. It is important for prescribers to ask patients or their caregivers specifically about the development of new or increased gambling urges, sexual urges, compulsive spending, binge or compulsive eating, or other urges while being treated with aripiprazole. It should be noted that impulse-control symptoms can be associated with the underlying disorder; however, in some cases urges were reported to have stopped when the dose was reduced or the medication was discontinued. Impulse control disorders may result in harm to the patient and others if not recognized. Consider dose reduction or stopping the medication if a patient develops such urges while taking aripiprazole. See Section 4.8 Adverse Effects (Undesirable Effects), Post-market adverse drug reactions.
Weight gain.
Antipsychotic drugs have been associated with metabolic changes, including weight gain. In long-term clinical trials there was no clinically relevant difference in the incidence of weight gain between aripiprazole and placebo. In a 12-week clinical trial in acutely relapsed patients, a potentially clinically relevant difference in the incidence of weight gain between aripiprazole and placebo was seen. See Section 4.8 Adverse Effects (Undesirable Effects), Weight.
All patients should have baseline and periodic monitoring of body weight, and other cardiometabolic parameters, including fasting glucose, full lipid profile and blood pressure, during treatment with any atypical antipsychotic including aripiprazole.
Paediatric use.
The safety and efficacy of aripiprazole in children and adolescents aged 0-17 years have not been established. No data are available.
Use in the elderly.
There are no data on the safety and efficacy of aripiprazole in patients ≥ 61 years of age.
Effects on laboratory tests.
Drug interaction with laboratory tests has not been established.4.5 Interactions with Other Medicines and Other Forms of Interactions
While no specific drug interaction studies have been performed with aripiprazole, the effects of co-administration of inhibitors of CYP2D6 and CYP3A4 were modelled as part of a population pharmacokinetic study but with no data accrued. The information below is therefore obtained from studies with oral aripiprazole.
Potential for other medicinal products to affect aripiprazole.
Aripiprazole is metabolised by multiple pathways involving the CYP2D6 and CYP3A4 enzymes.
Inhibitors and inducers of CYP2D6 and CYP3A4. Quinidine or other strong CYP2D6 inhibitors.
In a clinical trial of oral aripiprazole in healthy subjects, a strong inhibitor of CYP2D6 (quinidine) decreased oral clearance of aripiprazole by 52%, increased aripiprazole AUC by 107%, while Cmax was unchanged. The AUC and Cmax of dehydro-aripiprazole, the active metabolite, decreased by 32% and 47%, respectively. Other strong inhibitors of CYP2D6, such as fluoxetine, paroxetine, and bupropion may be expected to have similar effects and similar dose reduction should, therefore, be applied. See Section 4.2 Dose and Method of Administration, Dosage in special populations, Dose adjustments due to interactions.
Ketoconazole or other strong CYP3A4 inhibitors.
In a clinical trial of oral aripiprazole in healthy subjects, a strong inhibitor of CYP3A4 (ketoconazole) decreased oral clearance of aripiprazole by 38%, increased aripiprazole AUC and Cmax by 63% and 37%, respectively. The AUC and Cmax of dehydro-aripiprazole increased by 77% and 43%, respectively. Other strong inhibitors of CYP3A4, such as itraconazole and HIV protease inhibitors may be expected to have similar effects and similar dose reductions should, therefore, be applied (see Section 4.2 Dose and Method of Administration, Dosage in special populations, Dose adjustments due to interactions). When considering concomitant administration of ketoconazole or other potent CYP3A4 inhibitors with aripiprazole, potential benefits should outweigh the potential risks to the patient.
Upon discontinuation of the CYP2D6 or CYP3A4 inhibitor, the dose of aripiprazole should be increased to the dose prior to the initiation of the concomitant therapy.
Carbamazepine or other CYP3A4 or CYP2D6 inducers.
In a clinical study in patients with schizophrenia or schizo-affective disorder, co-administration of carbamazepine (200 mg twice daily), a potent CYP3A4 inducer, with aripiprazole (30 mg daily) resulted in an approximate 70% decrease in AUC values of both aripiprazole and its active metabolite, dehydro-aripiprazole. Concomitant administration of aripiprazole and other potent inducers of CYP3A4 (such as rifampicin, rifabutin, phenytoin, phenobarbital, primidone, efavirenz, nevirapine and St. John's wort) or potent inducers of CYP2D6 may be expected to have similar effects.
Valproate and lithium.
When either valproate or lithium was administered concomitantly with aripiprazole, there was no clinically significant change in aripiprazole concentrations.
Inhibitors and inducers of CYP1A1, CYP1A2, CYP2C9 and CYP2C19. Aripiprazole is not metabolised by CYP1A1, CYP1A2, CYP2C9 and CYP2C19 in vitro, suggesting that interactions with medications or other factors (e.g. smoking), which are inhibitors or inducers of these enzymes, are unlikely.
Potential for aripiprazole to affect other medicinal products.
CNS drugs (including alcohol).
Given the primary CNS effects of aripiprazole, caution should be used when aripiprazole is administered in combination with alcohol or other CNS drugs with overlapping adverse reactions, such as sedation. See Section 4.8 Adverse Effects (Undesirable Effects).
Patients should be advised to avoid alcohol while on aripiprazole.
When aripiprazole was administered concomitantly with valproate, lithium, lamotrigine, dextromethorphan, warfarin, omeprazole, escitalopram, venlafaxine or desvenlafaxine there was no clinically important change in concentrations of these drugs.
Effects of aripiprazole on substrates for CYP2D6, CYP2C9, CYP2C19, CYP3A4 and CYP1A2.
In clinical studies, oral doses of 10-30 mg/day of aripiprazole had no significant effect on the metabolism of substrates of CYP2D6 (dextromethorphan/3-methoxymorphinan ratio), 2C9 (warfarin), 2C19 (omeprazole), and 3A4 (dextromethorphan). Additionally, aripiprazole and dehydro-aripiprazole did not show potential for altering CYP1A2-mediated metabolism in vitro. Thus, aripiprazole is unlikely to cause clinically important medicinal product interactions mediated by these enzymes.
Antihypertensive agents.
Due to its α1-adrenergic receptor antagonistic activity, aripiprazole has the potential to enhance the effect of certain antihypertensive agents.
Medicines which cause QT prolongation or electrolyte imbalance.
If aripiprazole is administered concomitantly with medicinal products known to cause QT prolongation or electrolyte imbalance, caution should be used.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Reproductive toxicity studies have not been performed on aripiprazole. The following information is taken from studies performed on oral aripiprazole, which showed that aripiprazole did not impair fertility in reproductive toxicity studies.
Aripiprazole had no effect on fertility in female rats treated orally with 2, 6, and 20 mg/kg/day (0.6, 2, and 6 times the oral maximum recommended human dose (MRHD) of 30 mg/day based on mg/m2) for 2 weeks prior to mating through gestation day 7. Drug-related effects (persistent dioestrus, and increased mating time, pre-implantation losses, and corpora lutea) observed at all doses were considered the result of perturbed oestrous cyclicity secondary to drug-mediated hyperprolactinaemia.
Aripiprazole had no effect on fertility in male rats treated with oral doses of 20, 40, and 60 mg/kg/day (6, 12, and 18 times the oral MRHD of 30 mg/day based on mg/m2) for 9 weeks prior to mating through mating. Disturbances of spermatogenesis were seen at 60 mg/kg/day and prostatic atrophy was seen at 40 and 60 mg/kg/day.
(Category C)
There are no adequate and well-controlled studies of aripiprazole in pregnant women. It is not known whether aripiprazole can cause fetal harm when administered to a pregnant woman or can affect reproductive capacity. Patients must be advised to notify their doctors if they become pregnant or intend to become pregnant during treatment with aripiprazole.
In animal studies, aripiprazole demonstrated developmental toxicity, including possible teratogenic effects in rats and rabbits. Pregnant rats were treated with oral doses of 3, 10, and 30 mg/kg/day (1, 3, and 9 times the oral maximum recommended human dose [MRHD] of 30 mg/day on a mg/m2 basis) of aripiprazole during the period of organogenesis. At 30 mg/kg in the rat, treatment was associated with slightly prolonged gestation, and a slight delay in fetal development, as evidenced by decreased fetal weight, undescended testes, and delayed skeletal ossification. There were no adverse effects on embryofetal or pup survival. Delivered offspring had decreased bodyweights, and increased incidences of hepatodiaphragmatic nodules and diaphragmatic hernia at 30 mg/kg (the other dose groups were not examined for these findings). A low incidence of diaphragmatic hernia was also seen in the fetuses exposed to 30 mg/kg. Postnatally, delayed vaginal opening was seen at 10 and 30 mg/kg and impaired reproductive performance (decreased fertility rate, corpora lutea, implants, and live fetuses and increased post implantation loss, likely mediated through effects on female offspring) was seen at 30 mg/kg. Some maternal toxicity was seen at 30 mg/kg; however, there was no evidence to suggest that these developmental effects were secondary to maternal toxicity.
Pregnant rabbits were treated with oral doses of 10, 30, and 100 mg/kg/day (2, 3, and 11 times human exposure at the oral MRHD of 30 mg/day based on AUC) of aripiprazole during the period of organogenesis. Decreased maternal food consumption and increased abortions were seen at 100 mg/kg. Treatment caused increased fetal mortality (100 mg/kg), decreased fetal weight (30 and 100 mg/kg), increased incidence of a skeletal abnormality (fused sternebrae at 100 mg/kg), and minor skeletal variations (100 mg/kg).
Rats were treated with oral doses of 3, 10, and 30 mg/kg/day (1, 3, and 9 times the oral MRHD of 30 mg/day on a mg/m2 basis) of aripiprazole from late gestation through weaning. At 30 mg/kg, maternal toxicity, slightly prolonged gestation, an increase in stillbirths, poor postnatal care/nursing, and decreases in pup weight (persisting into adulthood) and survival were seen.
Non-teratogenic class effect.
Neonates exposed to antipsychotic drugs (including aripiprazole) during the third trimester of pregnancy are at risk of experiencing extrapyramidal neurological disturbances and/or withdrawal symptoms following delivery. There have been post-marketing reports of agitation, hypertonia, hypotonia, tremor, somnolence, respiratory distress, and feeding disorder in these neonates. These complications have varied in severity; while in some cases symptoms have been self-limited; in other cases neonates have required additional medical treatment or monitoring.
Aripiprazole should be used during pregnancy only if the anticipated benefit to the mother outweighs the potential risk to the fetus and the administered dose and duration of treatment should be as low and as short as possible.
Use in labour and delivery.
The effect of aripiprazole on labour and delivery in humans is unknown.
Aripiprazole has been found in human breast milk. Patients should be advised not to breastfeed if they are taking aripiprazole.
In rats, there were adverse effects in dams and offspring following daily oral administration of aripiprazole from late gestation through weaning. See Section 4.6 Fertility, Pregnancy and Lactation, Use in pregnancy.4.7 Effects on Ability to Drive and Use Machines
Aripiprazole, like other antipsychotics, may have the potential to impair judgment, thinking, or motor skills. Patients should be cautioned about operating hazardous machinery, including motor vehicles, until they are reasonably certain that aripiprazole therapy does not affect them adversely.
The effects of this medicine on a person's ability to drive and use machines were not assessed as part of its registration.
4.8 Adverse Effects (Undesirable Effects)
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.
Aripiprazole IM depot administered once monthly has been evaluated for safety in 3,453 adult patients in clinical trials in schizophrenia and bipolar I disorder. Of the 3,453 adult patients exposed to aripiprazole IM depot, 2,567 patients have been treated with aripiprazole IM depot 400 mg/300 mg. Of the 3,453 patients exposed to aripiprazole IM depot 1,226 patients have received at least 13 aripiprazole 400 mg/300 mg injections (i.e. have been treated for at least 12 months).
Aripiprazole has been evaluated for safety in 13,543 patients who participated in multiple dose clinical trials across all approved indications including schizophrenia and bipolar I disorder, and who had approximately 7619 patient-years of exposure to oral aripiprazole and 749 patients with exposure to aripiprazole short acting injection. A total of 3390 patients were treated with oral aripiprazole for at least 180 days and 1933 patients treated with oral aripiprazole had at least 1 year of exposure.
Adult patients with schizophrenia.
Aripiprazole IM depot administered once monthly has been evaluated for safety in clinical trials in adult patients with schizophrenia. Of the 2,649 adult patients exposed to aripiprazole long-acting injectable, 2,567 patients have been treated with aripiprazole IM depot 400/300 mg.
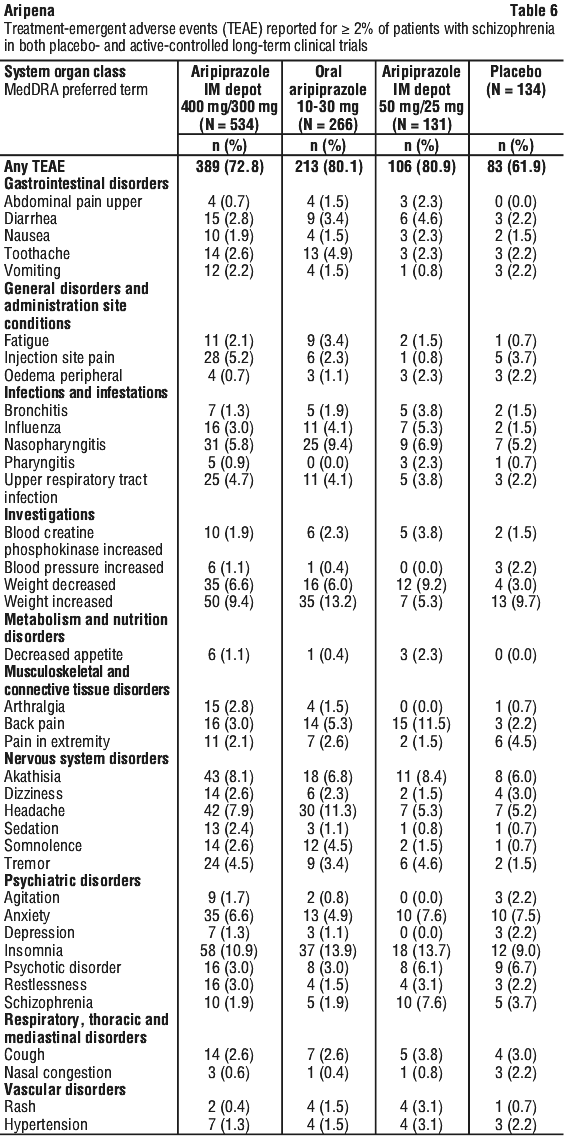
The most frequently observed treatment-emergent adverse events (TEAEs) reported in ≥ 5% of patients treated with aripiprazole IM depot 300-400 mg in the two double-blind long-term clinical trials were insomnia (10.9%), weight increased (9.4%), akathisia (8.1%), headache (7.9%), anxiety (6.6%), decreased weight (6.6%), nasopharyngitis (5.8%), and injection site pain (5.2%). Overall, treatment emergent adverse events (TEAEs) were similar to placebo and the majority were mild to moderate in severity. The TEAEs that occurred in the two double-blind long-term clinical trials with aripiprazole at a frequency of ≥ 2% are listed in Table 6.
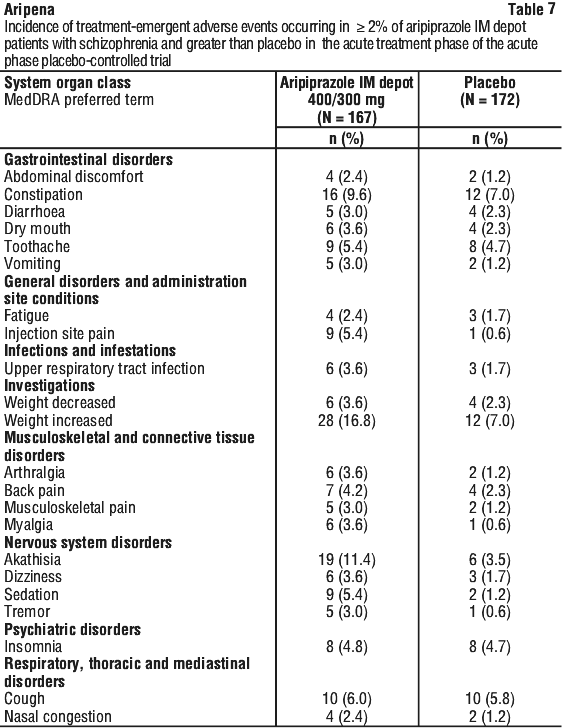
 The most frequently observed treatment emergent adverse event (TEAE) reported in ≥ 5% of patients treated with aripiprazole IM depot in the 12-week clinical trial in acutely relapsed patients were weight increased (16.8%), headache (14.4%), akathisia (11.4%), constipation (9.6%), cough (6.0%), dyspepsia (6.0%), agitation (5.4%), injection site pain (5.4%), sedation (5.4%) and toothache (5.4%). The TEAEs that occurred in at least 2% of patients and greater than placebo during the active treatment phase of the 12-week clinical trial in patients in the acute phase of schizophrenia are listed in Table 7.
The most frequently observed treatment emergent adverse event (TEAE) reported in ≥ 5% of patients treated with aripiprazole IM depot in the 12-week clinical trial in acutely relapsed patients were weight increased (16.8%), headache (14.4%), akathisia (11.4%), constipation (9.6%), cough (6.0%), dyspepsia (6.0%), agitation (5.4%), injection site pain (5.4%), sedation (5.4%) and toothache (5.4%). The TEAEs that occurred in at least 2% of patients and greater than placebo during the active treatment phase of the 12-week clinical trial in patients in the acute phase of schizophrenia are listed in Table 7.
Adult patients with bipolar I disorder.
Aripiprazole IM depot administered once monthly has been evaluated for safety in clinical trials in 804 adult patients with bipolar I disorder.
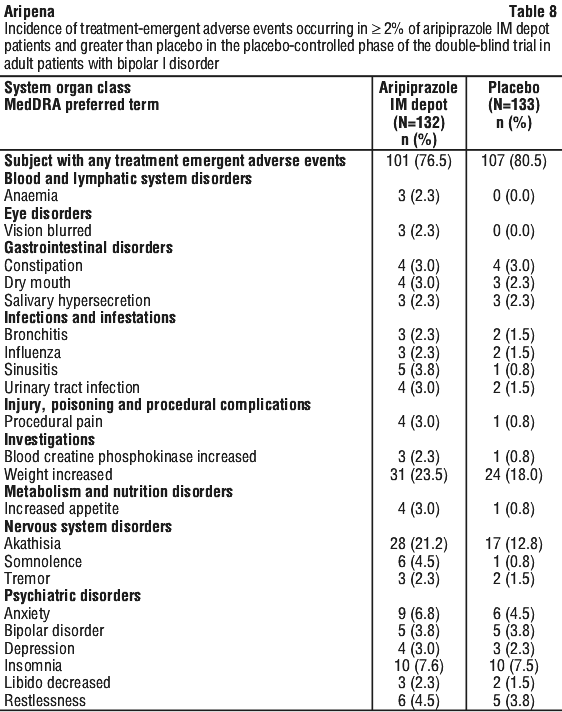
In a double-blind, placebo-controlled, randomized withdrawal (maintenance) study, the most frequently observed TEAEs that were reported in ≥ 5 % of patients receiving aripiprazole IM depot and with a greater frequency than in the placebo group were weight increased (23.5%), akathisia (21.2%), insomnia (7.6%) and anxiety (6.8%). See Table 8.
Injection site adverse events.
Injection site assessments were completed after all injections during the double-blind, controlled phases of the two aripiprazole IM depot long-term trials and the acute schizophrenia trial. Analyses of injection site assessments (investigator-rated and subject reported Visual Analogue Scale (VAS)) were performed to evaluate the safety/tolerability of aripiprazole IM depot, see Table 9. In both long-term trials, infrequent injection site reactions were observed; those seen were generally mild to moderate in severity, and resolved over time. Injection site pain (incidence 5.2 %), has a median onset on day 2 after the injection and a median duration of 4 days. In the acute phase trial, injection site pain (incidence 5.4%) was reported to be mild in severity, and resolved over time.
In the double-blind, placebo-controlled phase in patients with bipolar I disorder, the percentage of patients reporting injection site-related adverse reactions was similar between aripiprazole IM depot and placebo. The mean intensity of injection pain reported by patients on aripiprazole IM depot using a visual analog scale (0=no pain to 100=unbearably painful) was 5.2 (SD 9.0) for the first injection and 4.0 (SD 7.6) for the last injection in the double-blind, placebo-controlled phase.
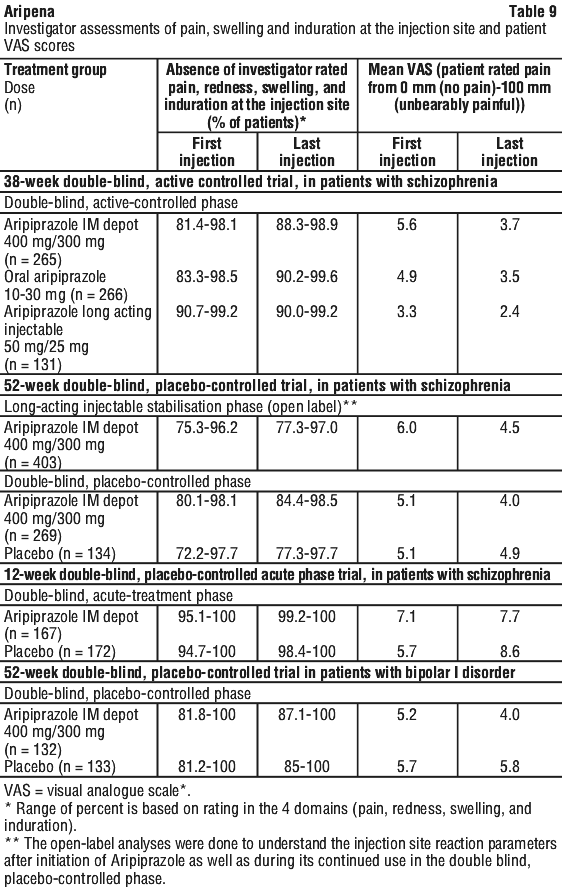 In an open label study comparing the bioavailability of aripiprazole IM depot administered in the deltoid or gluteal muscle, injection site-related reactions were slightly more frequent in the deltoid muscle. The majority were mild and improved on subsequent injections. Repeated occurrence of injection site pain was more frequent in the deltoid muscle.
In an open label study comparing the bioavailability of aripiprazole IM depot administered in the deltoid or gluteal muscle, injection site-related reactions were slightly more frequent in the deltoid muscle. The majority were mild and improved on subsequent injections. Repeated occurrence of injection site pain was more frequent in the deltoid muscle.
Leukopenia.
Neutropenia has been reported in the clinical program with aripiprazole and typically starts around day 16 after first injection, and lasts a median of 18 days.
Extrapyramidal symptoms (EPS).
During the double-blind phases of the 38- and 52-week trials in schizophrenia, treatment-emergent EPS and EPS-related events were reported for aripiprazole IM depot (18.4%) and oral aripiprazole tablets 10-30 mg (11.7%). The most commonly reported EPS and EPS-related events in each group were akathisia events (aripiprazole IM depot 300/400 mg: 8.2%; oral aripiprazole tablets 10-30 mg: 6.8%; and placebo: 6.0%), followed by parkinsonism events (6.9%; 4.1% and 3.0, respectively). Akathisia typically starts around day 10 after first injection, and lasts a median of 56 days.
There was minimal variation in EPS symptoms during the double-blind phases as assessed by mean changes from baseline in the Simpson-Angus Scale (SAS), Abnormal Involuntary Movement Scale (AIMS) and Barnes Akathisia Rating Scale (BARS) rating scales. The mean changes were not considered to be clinically relevant.
During the double-blind, placebo-controlled phase of the bipolar I disorder trial, 36/132 (27.3%) aripiprazole IM depot-treated subjects and 22/133 (16.5%) placebo-treated subjects had treatment-emergent EPS and EPS-related AEs. In the aripiprazole (vs. placebo) group, EPS and EPS-related events reported by ≥ 2% of subjects were akathisia and psychomotor hyperactivity events (22.0% vs 12.8%), parkinsonism events (5.3% vs 3.8%), dyskinetic (2.3% vs 1.5%), and dystonic events (2.3% vs 0.0%).
The most frequently reported treatment-emergent EPS and EPS-related AE was akathisia with 28/132 (21.2%) aripiprazole-treated subjects and 17/133 (12.8%) placebo-treated subjects experiencing an event. One aripiprazole-treated and no placebo-treated subjects experienced an SAE of akathisia and 2 aripiprazole-treated subjects were discontinued due to akathisia. There were no other EPS-related SAEs and no other TEAEs leading to discontinuation reported.
Dystonia.
Class effect.
Symptoms of dystonia, prolonged abnormal contractions of muscle groups, may occur in susceptible individuals during the first few days of treatment. Dystonic symptoms include: spasm of the neck muscles, sometimes progressing to tightness of the throat, swallowing difficulty, difficulty breathing, and/or protrusion of the tongue. While these symptoms can occur at low doses, they occur more frequently and with greater severity with high potency and at higher doses of first generation antipsychotic drugs. An elevated risk of acute dystonia is observed in males and younger age groups.
Weight.
During the double-blind, active-controlled phase of the 38-week trial in schizophrenia, the incidence of weight gain of ≥ 7% from baseline to last visit was 9.5% for the aripiprazole IM depot group and 11.7% for the oral aripiprazole tablets 10-30 mg group. The incidence of weight loss of ≥ 7% from baseline to last visit was 10.2% for aripiprazole IM depot and 4.5% for oral aripiprazole tablets 10-30 mg. During double-blind treatment, mean change in body weight from baseline to last visit was -0.2 kg for aripiprazole IM depot, +0.7 kg for oral aripiprazole tablets.
During the double-blind, placebo-controlled phase of the 52-week aripiprazole IM depot schizophrenia trial, the incidence of weight gain of ≥ 7% from baseline to last visit was similar between aripiprazole and placebo: 6.4% for the aripiprazole group and 5.2% for the placebo group. The incidence of weight loss of ≥ 7% from baseline to last visit was 6.4% for the aripiprazole IM depot group and 6.7% for the placebo group. During double-blind treatment, mean change in body weight from baseline to last visit was -0.2 kg for aripiprazole and -0.4 kg for placebo (p=0.812).
In the 12-week clinical trial in acutely relapsed schizophrenia patients, the incidence of potentially clinically relevant weight gain of ≥ 7% from baseline to last visit was 21.5% for the aripiprazole group and 8.5% for the placebo group. The mean change in body weight from baseline to last visit was +2.8 kg (N=144) for aripiprazole and +0.8 kg (N=141) for placebo with a median exposure of 85 days.
During the double-blind, placebo-controlled phase of the bipolar I disorder trial, the incidence of weight gain ≥ 7% at any time was 18.0% in the aripiprazole group and 12.9% for the placebo group; the incidence of weight loss ≥ 7% at any time was 9.4% in the aripiprazole group and 12.1% in the placebo group. At the last visit, the incidence of potentially clinically relevant weight gain was 13.3% in the aripiprazole group and 12.1% in the placebo group; the incidence of weight loss ≥ 7% at last visit was 5.5% in the aripiprazole group and 10.6% in the placebo group. The mean change (SD) from baseline at week 52 was 1.3 (5.9) kg for the aripiprazole group and 1.5 (6.1) kg for the placebo group; at the last visit, the mean change from baseline was 0.9 (5.3) kg for the aripiprazole group and 0.0 (5.9) kg for the placebo group.
Patients should have baseline and periodic monitoring of body weight, and other cardiometabolic parameters, including fasting glucose, full lipid profile and blood pressure, during treatment with any atypical antipsychotic including aripiprazole.
Laboratory tests.
No clinically relevant mean changes from baseline in serum chemistry, haematology, urinalysis or other laboratory test (e.g. insulin, fasting insulin) results were observed during the acute or long-term clinical trials with aripiprazole IM depot.
Comparisons between oral aripiprazole and placebo in the proportions of patients experiencing potentially clinically significant changes in routine laboratory and lipid parameters revealed no medically important differences. Elevations of CPK (creatine phosphokinase), generally transient and asymptomatic, were observed in 3.5% of patients treated with oral aripiprazole as compared to 2.0% of patients treated with placebo.
QT interval.
During double-blind treatment in the long-term trials, 1/534 (0.2%) aripiprazole IM depot subject had a treatment-emergent adverse event related to QT interval change (prolonged ECG QT).
Prolactin.
Changes in prolactin levels were comparable across trials, irrespective of indication (schizophrenia or bipolar I disorder), and there were no clinically relevant mean changes from baseline to the last visit with regard to prolactin levels during the double-blind treatment phase of either trial.
In the double-blind, active-controlled phase of the 38-week trial, from baseline to last visit there was a mean decrease in prolactin levels in the aripiprazole IM depot group (-0.33 nanogram/mL) compared with a mean increase in the oral aripiprazole tablets 10-30 mg group (0.79 nanogram/mL; p < 0.01). The incidence for aripiprazole IM depot patients with prolactin levels > 1 time the upper limit of normal (ULN) range at any assessment was 5.4% compared with 3.5% of oral aripiprazole tablets 10-30 mg, with a higher incidence in male patients than female patients in each treatment group.
In the double-blind, placebo-controlled phase of the 52-week trial in schizophrenia, from baseline to last visit, there was a mean decrease in prolactin levels in the aripiprazole IM depot group (-0.38 nanogram/mL) compared with a mean increase in the placebo group (1.67 nanogram/mL). The incidence of aripiprazole patients with prolactin levels > 1 time the ULN was 1.9% compared to 7.1% for placebo patients.
Of note, differences in the mean (± SD) change from the double-blind treatment phase baseline to the last visit of the double-blind treatment phase in prolactin were negligible between the aripiprazole and placebo groups and of little if any clinical relevance, indicating no implications for adverse effects on prolactin.
In the double-blind, placebo-controlled phase of the bipolar I disorder trial, the mean changes from baseline to last visit in prolactin were minimal in the aripiprazole IM depot 400 mg/300 mg group (0.15 nanogram/mL) compared to placebo (3.00 nanogram/mL) and none of the changes were considered to be clinically meaningful. There were no clinically meaningful differences in the incidence of prolactin levels above the ULN between treatment groups and no incidence of > 3 x ULN reported during the double-blind, phase of this trial. No clinically meaningful differences in shifts in prolactin between treatment groups or gender were reported. During the double-blind phase, no aripiprazole-treated and 0.8% of placebo-treated subjects experienced the prolactin-related TEAE of hyperprolactinaemia.
Lipid parameters.
In the two double-blind studies of 38 and 52 weeks' duration, the differences in the mean change from baseline (double-blind treatment phase) to the last visit in fasting lipid parameter values (total cholesterol, triglycerides, HDL, and LDL) were negligible between the aripiprazole IM depot 400 mg/300 mg group compared with oral aripiprazole tablets 10‑30 mg group, aripiprazole IM depot 50 mg/25 mg group or placebo groups and could be considered as of no major clinical concern.
Other adverse reactions observed during the clinical trial evaluation of aripiprazole.
All reported events in the aripiprazole IM depot group during the randomisation phase of the clinical trials, reported by less than 2% of subjects, and at least as frequently as in the placebo group are listed below.
Blood and lymphatic system disorders.
Anaemia, bicytopenia, leukopenia, lymphadenopathy, neutropenia, thrombocytopenia.
Cardiac disorders.
Acute myocardial infarction, atrial fibrillation, bradycardia, cardio-respiratory arrest, first degree atrioventricular blocks, cardiac failure congestive, ventricular extrasystoles.
Ear and labyrinth disorders.
Deafness, tinnitus, vertigo.
Eye disorders.
Conjunctivitis allergic, eye irritation, eye pain, eyelid ptosis, oculogyric crisis, photophobia, vision blurred.
Gastrointestinal disorders.
Abdominal distension, abdominal pain, abdominal pain upper, anorectal discomfort, aphthous stomatitis, colitis, constipation, dental caries, diverticulum, dry mouth, dyspepsia, dysphagia, food poisoning, frequent bowel movements, gastritis, gastroesophageal reflux disease, gingival oedema, gingival pain, gingivitis, haemorrhoidal haemorrhage, haemorrhoids, inguinal hernia, loose tooth, nausea, oral discomfort, periodontitis, poor dental condition, salivary hypersecretion, tongue disorder, tooth impacted, tooth loss, vomiting.
General disorders and administration site conditions.
Asthenia, chest discomfort, gait disturbance, influenza-like illness, infusion site haematoma, infusion site swelling, injection related reaction, injection site discomfort, injection site pruritus, injection site induration, injection site pain, injection site reaction, injection site swelling, lethargy, night sweats, oedema peripheral, pain, sluggishness, suprapubic pain, swollen tongue, thirst, vessel puncture site haematoma, vessel puncture site pain.
Hepatobiliary disorders.
Cholecystitis chronic, cholelithiasis, hepatic cirrhosis, hepatic steatosis, hepatosplenomegaly.
Immune system disorders.
Drug hypersensitivity.
Infections and infestations.
Acarodermatitis, anal abscess, appendicitis perforated, breast cellulitis, chlamydial infection, cellulitis, cystitis, dermatitis, ear infection, Escherichia UTI, folliculitis, fungal infection, fungal skin infection, furuncle, gastroenteritis, gastroenteritis viral, herpes virus infection, herpes zoster, hordeoleum, impetigo, laryngitis, lice infestation, localised infection, mastitis, oral candidiasis, oropharyngitis fungal, otitis externa, otitis media, pharyngitis, pharyngitis streptococcal, pilonidal cyst, pneumonia, respiratory tract infection, sycosis barbae, trichomoniasis, viral rhinitis, subcutaneous abscess, tinea pedis, tooth abscess, tooth infection, urinary tract infections, vaginal infection, varicella, viral infection, viral upper respiratory tract infection, vulvovaginal mycotic infection.
Injury, poisoning and procedural complications.
Accident, ankle fracture, arthropod bite, carbon monoxide poisoning, chest injury, contusion, drug toxicity, excoriation, face injury, fall, foot fracture, gunshot wound, injury, joint dislocation, joint sprain, laceration, multiple injuries, muscle injury, muscle oedema, muscle strain, procedural pain, radius fracture, skeletal injury, skin laceration, spinal column injury, thermal burn, tooth fracture, wound.
Investigations.
Alkaline phosphatase increased, bilirubin increased, blood creatinine phosphokinase increased, blood insulin increased, cholesterol decreased, glucose decreased, glucose increased, lactate dehydrogenase increased, triglycerides decreased, triglycerides increased, electrocardiogram abnormal, electrocardiogram QT prolonged, electrocardiogram ST segment depression, electrocardiogram T wave amplitude decreased, electrocardiogram T wave inversion, gamma-glutamyltransferase increased, glucose urine present, glycosylated haemoglobin increased, heart rate decreased, hepatic enzyme increased, liver function test abnormal, liver function test increased, neutrophil count decreased, protein urine, waist circumference increased, white blood cell count decreased, white blood cells urine.
Metabolism and nutrition disorders.
Appetite disorder, decreased appetite, diabetes mellitus, gout, hypercholesterolaemia, hyperglycaemia, hyperinsulinaemia, hyperlipidaemia, hypertriglyceridaemia, hyperuricaemia, hypoglycaemia, increased appetite, overweight, type 2 diabetes mellitus.
Musculoskeletal and connective tissue disorders.
Arthritis, joint swelling, muscle rigidity, muscle haemorrhage, muscle spasm, muscle tightness, muscle twitching, musculoskeletal pain, myalgia, neck pain, nuchal rigidity, rotator cuff syndrome, sciatica, trismus.
Neoplasms benign malignant and unspecified.
Basal cell carcinoma, breast fibroma, pancreatic carcinoma.
Nervous system disorders.
Bradykinesia, brain injury, cogwheel rigidity, disturbance in attention, drooling, dyskinesia, dystonia, extrapyramidal disorder, hypersomnia, hypoaesthesia, migraine, movement disorder, paraesthesia, parkinsonism, parosmia, poor quality sleep, post-traumatic neck syndrome, psychomotor hyperactivity, restless leg syndrome, sedation, sinus headache, syncope, tardive dyskinesia, tension headache, dizziness, transient ischaemic attack.
Psychiatric disorders.
Abnormal dreams, affect lability, apathy, bruxism, bulimia nervosa, delusion, dysphemia, dysphoria, hallucination auditory, hypersexuality, hypomania, hyposomnia, initial insomnia, irritability, libido decreased, middle insomnia, mood altered, nightmare, panic attack, panic reaction, psychomotor retardation, sleep disorder, social avoidant behaviour, somnambulism, suicidal ideation, suicide attempt, tension.
Renal and urinary disorders.
Asymptomatic bacteriuria, glycosuria, hypertonic bladder, micturition urgency, nephrolithiasis, pollakiuria.
Reproductive system and breast disorders.
Adnexa uteri pain, breast mass, breast tenderness, ejaculation delayed, erectile dysfunction, galactorrhoea, gynaecomastia, menorrhagia, ovarian cyst, sexual dysfunction, vulvovaginal dryness.
Respiratory thoracic and mediastinal disorders.
Allergic sinusitis, acute respiratory distress syndrome, asthma, dysphonia, dyspnoea, epistaxis, nasal septum deviation, non-cardiac chest pain oropharyngeal pain, paranasal sinus hypersecretion, respiratory failure, respiratory tract congestion, rhinalgia, rhinitis allergic, sinus congestion, wheezing, hiccups.
Skin and subcutaneous tissue disorders.
Acne, blister, dermatitis contact, dry skin, eczema, erythema, hyperkeratosis, pityriasis, pruritus, psoriasis, rash, rash macula, rosacea, skin induration, skin lesion, skin striae, urticaria.
Social circumstances.
Poor personal hygiene.
Vascular disorders.
Orthostatic hypertension.
Post-market adverse drug reactions.
The following adverse reactions have been reported during post-marketing surveillance with aripiprazole. The frequency of these reactions cannot be estimated from available post-marketing data and the causal relationship to the drug cannot be definitely established in the post-marketing scenario.
Blood and lymphatic system disorders.
Leukopenia, neutropenia, thrombocytopenia, blood prolactin decrease.
Endocrine disorders.
Hyperglycaemia, diabetes mellitus, diabetic ketoacidosis, diabetic hyperosmolar coma.
Metabolism and nutrition disorders.
Anorexia, hyponatraemia.
Psychiatric disorders.
Agitation, hypersexuality, pathological gambling, impulse-control disorders, obsessive-compulsive disorder, eating disorder.
Nervous system disorders.
Speech disorder, grand mal convulsion.
Eye disorders.
Diplopia.
Vascular disorders.
Syncope, hypertension.
Respiratory, thoracic and mediastinal disorders.
Aspiration pneumonia, hiccups.
Gastrointestinal disorders.
Pancreatitis, dysphagia, diarrhoea.
Hepato-biliary disorders.
Jaundice, hepatitis.
Skin and subcutaneous tissue disorders.
Allergic reaction (e.g. anaphylactic reaction, angioedema, pruritus, or urticaria, rash, laryngospasm), hyperhidrosis, alopecia, drug reaction with eosinophilia and systemic symptoms (DRESS).
Musculoskeletal and connective tissue disorders.
Rhabdomyolysis, myalgia, musculoskeletal stiffness.
Renal and urinary disorders.
Urinary incontinence, urinary retention.
Reproductive system and breast disorders.
Priapism.
General disorders and administration site conditions.
Temperature regulation disorder (e.g. hypothermia, pyrexia), chest pain.
Investigations.
Blood creatine phosphokinase increased, blood glucose increased, blood glucose fluctuation, glycosylated haemoglobin increased, weight increased, weight decreased, alanine aminotransferase increased, aspartate aminotransferase increased, gamma-glutamyltransferase increased.
Although a causal relationship has not been established, cases of suicide attempt, suicidal ideation, and completed suicide, have been reported post marketing.
Undesirable effects known to be associated with antipsychotic medication which have also been reported in association with aripiprazole are neuroleptic malignant syndrome, tardive dyskinesia, and seizure.
Uncommon occurrences of depression and tachycardia have also been reported in association with aripiprazole.4.9 Overdose
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).
No cases of overdose associated with adverse reactions were reported in clinical studies with aripiprazole IM depot. Care must be taken to avoid inadvertent injection of this medicinal product into a blood vessel. Following any confirmed or suspected accidental overdose/inadvertent intravenous administration, close observation of the patient is needed and if any potentially medically serious sign or symptom develops, monitoring, which should include continuous electrocardiographic monitoring, is required. The medical supervision and monitoring should continue until the patient recovers.
A simulation of dose dumping showed that the predicted median aripiprazole concentration reaches a peak of 4,500 nanogram/mL which corresponds to approximately 9 times the upper therapeutic range. In case of dose dumping, aripiprazole concentrations are predicted to descend rapidly to the upper limit of the therapeutic window after approximately 3 days. By the 7th day, the median aripiprazole concentrations further decline to concentrations following an IM depot dose with no dose dumping. While overdose is less likely with parenteral than oral medicinal products, reference information for oral aripiprazole overdose is presented below.
Signs and symptoms.
In clinical trials and post-marketing experience, accidental or intentional acute overdose of aripiprazole alone was identified in adult patients with reported estimated doses up to 1,260 mg (42 times higher than the recommended daily aripiprazole dose, 30 mg) with no fatalities. The potentially medically important signs and symptoms observed included lethargy, increased blood pressure, somnolence, tachycardia, nausea, vomiting and diarrhoea. In addition, reports of accidental overdose with aripiprazole alone (up to 195 mg) in children have been received with no fatalities. The potentially medically serious signs and symptoms reported included somnolence, transient loss of consciousness and extrapyramidal symptoms.
Management of overdose.
Management of overdose should concentrate on supportive therapy, maintaining an adequate airway, oxygenation and ventilation, and management of symptoms. The possibility of multiple medicinal product involvement should be considered. Therefore, cardiovascular monitoring should be started immediately and should include continuous electrocardiographic monitoring to detect possible arrhythmias. Following any confirmed or suspected overdose with aripiprazole, close medical supervision and monitoring should continue until the patient recovers.
Haemodialysis.
Although there is no information on the effect of haemodialysis in treating an overdose with aripiprazole, haemodialysis is unlikely to be useful in overdose management, since aripiprazole is highly bound to plasma proteins.5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
The mechanism of action of aripiprazole, as well as other drugs having efficacy in schizophrenia and bipolar disorder, is unknown. It has been proposed that aripiprazole's efficacy is mediated through a combination of partial agonism at dopamine D2 and serotonin 5-HT1A receptors and antagonism at serotonin 5-HT2A receptors.
Aripiprazole IM depot activity is primarily due to the parent drug, aripiprazole. Aripiprazole exhibited antagonist properties in animal models of dopaminergic hyperactivity and agonist properties in animal models of dopaminergic hypoactivity. Aripiprazole exhibited high binding affinity in vitro for dopamine D2 and D3, serotonin 5-HT1A and 5-HT2A receptors (Ki values of 0.3, 0.8, 1.7, and 3.4 nanoM, respectively), and moderate affinity for dopamine D4, serotonin 5-HT2C and 5-HT7, α1-adrenergic, and histamine H1 receptors (Ki values of 44, 15, 39, 57, and 61 nanoM, respectively). Aripiprazole also exhibited moderate binding affinity for the serotonin reuptake site (Ki value of 98 nanoM) but no appreciable affinity for muscarinic receptors (IC50 > 1000 nanoM).
The predominant metabolite in human plasma, dehydro-aripiprazole, has been shown to have a similar affinity for dopamine D2 and D3 receptors (Ki values 0.4 and 0.5 nanoM, respectively) as the parent compound and a much lower affinity for the other receptor subtypes (serotonergic, noradrenergic and histaminergic receptors).
Interaction with receptors other than dopamine and serotonin subtypes may explain some of the other clinical effects of aripiprazole.
Aripiprazole oral doses ranging from 0.5 to 30 mg administered once a day to healthy subjects for 2 weeks produced a dose-dependent reduction in the binding of 11C-raclopride, a D2 receptor ligand, to the caudate and putamen detected by positron emission tomography.
Clinical trials.
Schizophrenia. The efficacy and safety of aripiprazole in the treatment of adult patients with schizophrenia was established in one pivotal short-term, randomised, double-blind, placebo-controlled trial in acutely relapsed patients and one pivotal long-term, randomised, double-blind, placebo-controlled trial.
Clinical efficacy in the acute phase of schizophrenia.
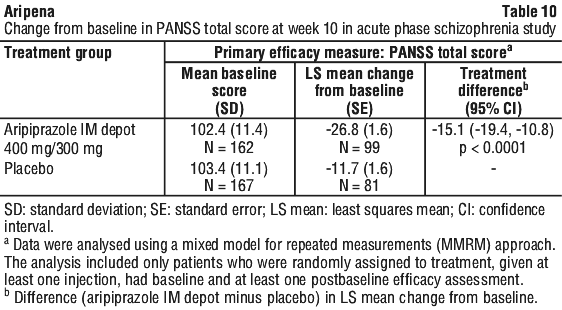
The efficacy of aripiprazole IM depot in adult patients in the acute phase of schizophrenia was established in one short-term (12 weeks), randomised, double-blind, placebo-controlled trial. Patients included in this trial met DSM-IV-TR criteria for schizophrenia and must have experienced an acute psychotic episode as defined by both a PANSS total score ≥ 80 and a PANSS score > 4 on each of four specific psychotic symptoms (conceptual disorganisation, hallucinatory behaviour, suspiciousness/persecution, unusual thought content) at baseline. Patients experiencing their first psychotic episode and those considered treatment resistant were excluded. Patients had a mean PANSS total score of 103 (range 82 to 144) and a CGI-S score of 5.2 (markedly to severely ill) at entry.
In this study patients were administered aripiprazole IM depot (n=167) or IM placebo (n=172) on days 0, 28, and 56. The dose could be adjusted down and up within the range of 400 mg to 300 mg on a one-time basis. Patients who had not taken aripiprazole previously had tolerability with oral aripiprazole (10 mg daily for 3 days) established prior to initiating treatment with aripiprazole IM depot or placebo. Patients randomised to aripiprazole IM depot also received concomitant oral aripiprazole, 10 to 20 mg/day, for the first two weeks of the study.
In the aripiprazole IM depot group, for 96.4% of patients, there was no difference between the starting dose and ending dose of aripiprazole IM depot (400 mg).
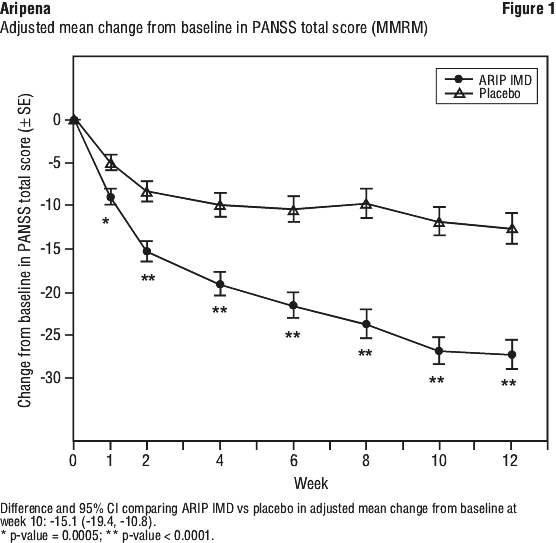
The primary endpoint was the change from baseline to week 10 in PANSS total score. Aripiprazole IM depot was superior to placebo in improving the PANSS total score, with week 10 scores of -26.8 and -11.7, respectively (see Table 10). A statistically significant difference (p ≤ 0.0001) was seen at each measured time point beginning at week 1 and continuing through to study completion. The adjusted mean change in PANSS total score over time is shown in Figure 1.
 For the key secondary endpoint, the change from baseline to week 10 in CGI-S score, the treatment difference between the aripiprazole IM depot group (LS mean change -1.4) and the placebo group (LS mean change -0.6) was -0.8 (95% CI: -1.1, -0.6), which was statistically significant (p < 0.0001).
For the key secondary endpoint, the change from baseline to week 10 in CGI-S score, the treatment difference between the aripiprazole IM depot group (LS mean change -1.4) and the placebo group (LS mean change -0.6) was -0.8 (95% CI: -1.1, -0.6), which was statistically significant (p < 0.0001).
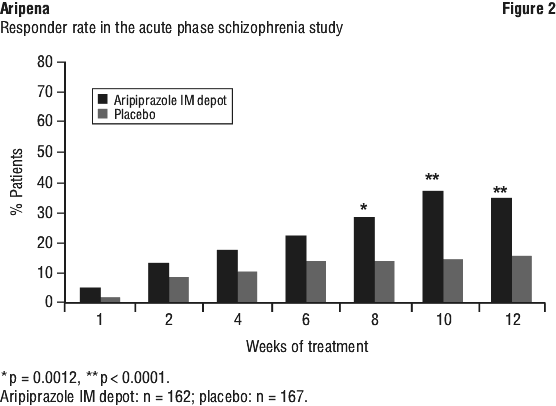
Response was defined as a ≥ 30% reduction from baseline in PANSS total score. The responder rate was numerically higher in the aripiprazole group at all post-baseline time points; the treatment differences were statistically significant (p ≤ 0.0013) from week 8 to week 12 (see Figure 2). At Week 10, the responder rate was 37.0% in the aripiprazole group compared to 14.4% in the placebo group; the treatment difference was 22.7% (95% CI 12.9%, 32.4%).
Clinical efficacy in the maintenance phase of schizophrenia.
The pivotal trial was a 52-week, randomised, double-blind, placebo-controlled trial conducted in adult patients with a current diagnosis of schizophrenia. This trial consisted of a screening phase and four treatment phases: conversion, oral stabilisation, aripiprazole IM depot stabilisation, and double-blind placebo-controlled. Patients eligible for the double-blind, placebo-controlled phase were randomly assigned in a 2:1 ratio to double-blind treatment with aripiprazole IM depot 400 mg (with an option to decrease to 300 mg for tolerability reasons) or placebo, respectively. The trial was completed early as a consequence of the positive pre-specified interim analysis and therefore only 26 patients completed 52 weeks of treatment. Eighty-seven per cent (87%) of subjects randomised to the 400 mg dose remained on this dose until either completing the trial duration or withdrawing from the trial.
The final efficacy analysis included 403 randomised patients and 80 exacerbations of psychotic symptoms/impending relapse events. The trial was terminated early because efficacy was demonstrated by the pre-specified interim analysis. The hazard ratio from the Cox proportional hazard model for the placebo to aripiprazole IM depot comparison was 5.029 (95% CI: 3.154, 8.018), thus patients in the placebo group had a 5-fold greater risk of experiencing impending relapse than patients in the aripiprazole IM depot group. The trial results support the efficacy for aripiprazole IM depot over 52 weeks of treatment.
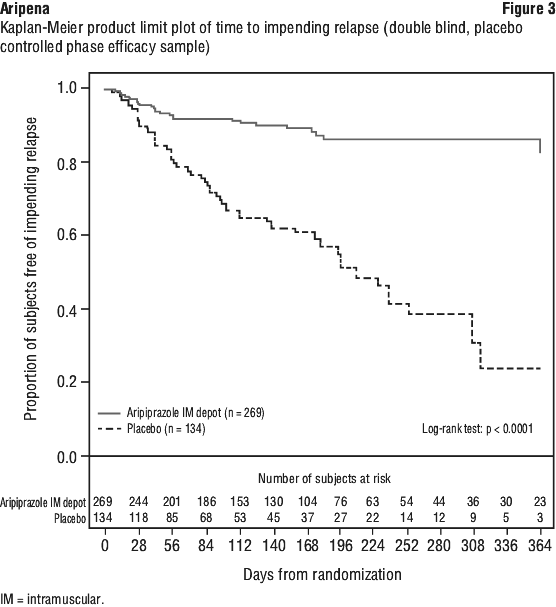
The Kaplan-Meier curves of the time from randomisation to impending relapse during the 52-week, double-blind treatment phase for aripiprazole and placebo groups are shown in Figure 3.
The percentage of patients meeting the impending relapse criteria was significantly lower (p < 0.0001) in the aripiprazole group (10.0%) than in the placebo group (39.6%).
The time to impending relapse was significantly shorter (p < 0.0001) for subjects in the placebo group compared with subjects in the aripiprazole group.
 Further, the superiority of aripiprazole IM depot compared to placebo is supported by the results of the analysis of PANSS total score, PANSS positive and negative subscales, CGIS, CGI-I. During the double blind phase, significant differences in mean PANSS total and CGI-S scores were observed.
Further, the superiority of aripiprazole IM depot compared to placebo is supported by the results of the analysis of PANSS total score, PANSS positive and negative subscales, CGIS, CGI-I. During the double blind phase, significant differences in mean PANSS total and CGI-S scores were observed.
Bipolar I disorder. Clinical efficacy in prevention of recurrence of manic or mixed episodes of bipolar 1 disorder.
The efficacy and safety of aripiprazole IM depot as treatment in adults aged 18 to 65 years was demonstrated in a 52-week, double-blind, placebo-controlled randomized withdrawal trial in patients who met DSM-IV-TR criteria for bipolar I disorder and who were experiencing a manic episode at trial entry. This trial consisted of a screening phase and 4 treatment phases: oral conversion, oral stabilization, aripiprazole IM depot stabilization, and a randomized, double-blind, placebo-controlled withdrawal phase.
Patients currently receiving oral treatment for their bipolar I disorder with medications other than aripiprazole monotherapy entered the oral conversion phase. In this phase, patients discontinued other treatments, such as mood stabilizers, antidepressants, or other antipsychotics over a period of 4 to 6 weeks and converted to aripiprazole monotherapy. Patients already treated with oral aripiprazole-monotherapy at the time of trial entry and those subjects converted to oral aripiprazole monotherapy in the oral conversion phase proceeded to the oral stabilization phase. Patients fulfilling the stabilization requirement were assigned to receive, in a single-blind fashion, aripiprazole IM depot 400 mg and began an IM depot stabilization phase for a minimum of 12 weeks and a maximum of 28 weeks. Patients who demonstrated stability for 8 consecutive weeks were randomized into the 52-week double-blind, placebo-controlled treatment phase. Of the 731 subjects who entered the trial, 466 subjects entered the conversion phase, 632 subjects entered the oral stabilization phase (including 265 subjects who entered the oral stabilization phase directly), and 425 subjects entered the IM depot stabilization phase. Stability was defined as: having achieved or maintained outpatient status; Young-Mania Rating Scale (YMRS) total score ≤ 12; Montgomery Asberg Depression Rating Scale (MADRS) total score ≤ 12 and no active suicidality; with active suicidality defined as a score of 4 or more on the MADRS Item 10 or an answer of "yes" on question 4 or 5 on the Columbia Suicide Severity Rating Scale (C-SSRS).
The final efficacy analysis included 265 patients and the primary efficacy endpoint is shown in the Kaplan-Meier curves of the cumulative proportion of patients with recurrence of any mood episode (manic, mixed or depressive) during the 52-week, double-blind treatment phase for aripiprazole IM depot and placebo treatment groups (see Figure 4). A total of 103 patients with recurrence of any mood episode were observed during the double-blind treatment phase: 35 occurred during aripiprazole treatment and 68 occurred during placebo treatment. The hazard ratio from the Cox proportional hazard model for the placebo to aripiprazole comparison was 2.220 (95% CI: 1.475, 3.340), thus patients receiving placebo had a 2.2-fold greater risk of experiencing a recurrence of a mood episode than patients receiving aripiprazole IM depot. These results support efficacy for aripiprazole IM depot over 52 weeks of treatment.
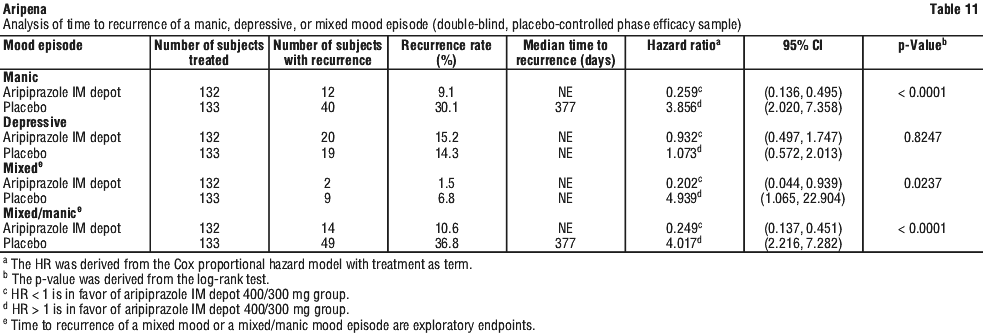
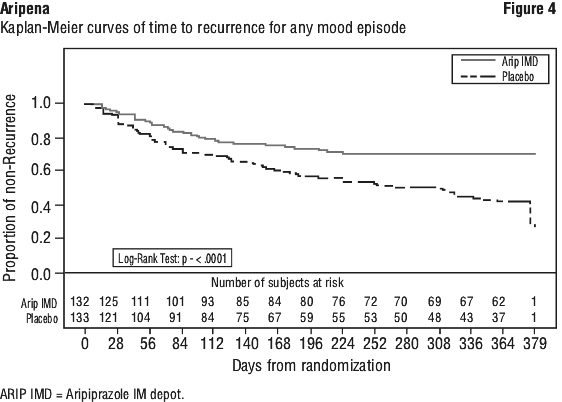 For the key secondary endpoint, the percentage of patients meeting the criteria for recurrence of any mood episode (manic, mixed, depressive) was significantly lower (Fisher's exact test p < 0.0001) in the aripiprazole group (26.5%) compared with placebo (51.1%). The times to recurrence of manic, mixed, and mixed/manic mood episodes were statistically significantly delayed in the aripiprazole IM depot group compared with the placebo group, with the log-rank test p-values of < 0.0001, 0.0237, and < 0.0001, respectively. The time to recurrence of a depressive mood episode showed no difference between the aripiprazole IM depot and placebo groups (p = 0.8247) (see Table 11).
For the key secondary endpoint, the percentage of patients meeting the criteria for recurrence of any mood episode (manic, mixed, depressive) was significantly lower (Fisher's exact test p < 0.0001) in the aripiprazole group (26.5%) compared with placebo (51.1%). The times to recurrence of manic, mixed, and mixed/manic mood episodes were statistically significantly delayed in the aripiprazole IM depot group compared with the placebo group, with the log-rank test p-values of < 0.0001, 0.0237, and < 0.0001, respectively. The time to recurrence of a depressive mood episode showed no difference between the aripiprazole IM depot and placebo groups (p = 0.8247) (see Table 11).
5.2 Pharmacokinetic Properties
Absorption.
Aripiprazole absorption into the systemic circulation is slow and prolonged following aripiprazole IM depot administration due to the low solubility of aripiprazole particles.
Following a single intramuscular dose of aripiprazole IM depot in the deltoid or gluteal muscle, the extent of absorption (AUC) is comparable for both injection sites, but the rate of absorption (Cmax) was higher following administration to the deltoid muscle.
Following multiple intramuscular doses, the plasma concentrations of aripiprazole gradually rise to a maximum plasma concentration at a median tmax of 4 days for the deltoid muscle and 7 days for the gluteal muscle.
Dose-proportional increases in aripiprazole and dehydro-aripiprazole concentrations and AUC parameters are observed after monthly aripiprazole IM depot injections of 400 mg and 300 mg. Steady state aripiprazole plasma concentrations were attained by the fourth monthly injection for both sites of administration.
Distribution.
Based on results from trials with oral administration of aripiprazole, aripiprazole is widely distributed throughout the body with an apparent volume of distribution of 4.9 L/kg, indicating extensive extravascular distribution. At therapeutic concentrations, aripiprazole is highly bound (88-97% to > 99%, as determined by polydimethylsiloxane-glass bead and equilibrium dialysis assays, respectively) to serum proteins, primarily albumin, in vitro. Aripiprazole did not alter the pharmacokinetics and pharmacodynamics of highly protein-bound warfarin, suggesting that protein displacement of warfarin did not occur.
Metabolism.
Aripiprazole undergoes minimal pre-systemic metabolism at the site of injection. Aripiprazole is extensively metabolised by the liver primarily by three biotransformation pathways: dehydrogenation, hydroxylation and N-dealkylation. Based on in vitro studies, CYP3A4 and CYP2D6 enzymes are primarily responsible for dehydrogenation and hydroxylation of aripiprazole, and N-dealkylation is catalysed by CYP3A4. Aripiprazole is the predominant drug moiety in systemic circulation. After multiple dose administration of aripiprazole IM depot, dehydro-aripiprazole, the active metabolite represents about 29.1-32.5% of aripiprazole AUC in plasma.
Excretion.
After administration of multiple doses of 400 mg or 300 mg of aripiprazole IM depot, the observed mean aripiprazole terminal elimination half-life is 46.5 and 29.9 days, respectively. Following a single oral dose of [14C]-labelled aripiprazole, approximately 27% of the administered radioactivity was recovered in the urine and approximately 60% in the faeces. Less than 1% of unchanged oral aripiprazole was excreted in the urine and approximately 18% was recovered unchanged in the faeces.
Special populations.
CYP2D6 poor metabolisers.
Based on population pharmacokinetic evaluation of aripiprazole IM depot, the total body clearance of aripiprazole was 3.71 L/h in extensive metabolisers of CYP2D6 (EMs) and approximately 1.88 L/h (approximately 50% lower) in poor metabolisers of CYP2D6 (PMs).
For dose recommendations see Section 4.2 Dose and Method of Administration, Dosage in special populations, Known CYP2D6 poor metabolisers. Subjects were entered into clinical studies without knowledge of their metaboliser status and, therefore, the safety profile reflects experience in both EMs and PMs.
Elderly patients.
After oral administration of aripiprazole, there are no clinically relevant differences in the pharmacokinetics of aripiprazole between healthy elderly and younger adults. Similarly, there was no detectable effect of age (18-61 year age range) in a population pharmacokinetic analysis of aripiprazole IM depot clinical trials in patients with schizophrenia.
Gender.
After oral administration of aripiprazole, there are no differences in the pharmacokinetics of aripiprazole between healthy male and female subjects when differences in body weight are considered. Population pharmacokinetic analysis of aripiprazole IM depot revealed a difference in the predicted mean half-lives between men (24 days) and women (32 days) as well as a gender dependent absorption rate. At steady state (model predicted) however, the parameters of Cmin, Cmax, and AUC0-tau did not exhibit any trends towards gender.
Race.
Population pharmacokinetic evaluation of aripiprazole showed no evidence of race-related differences in the pharmacokinetics of aripiprazole.
Renal impairment.
In a single-dose study with oral administration of aripiprazole, the pharmacokinetic characteristics of aripiprazole and dehydro-aripiprazole were found to be similar in patients with severe renal disease compared to those in young healthy subjects.
In patients with severe renal impairment (creatinine clearance < 30 mL/min), Cmax of aripiprazole (given in a single dose of 15 mg) and dehydro-aripiprazole increased by 36% and 53%, respectively, but AUC was 15% lower for aripiprazole and 7% higher for dehydro-aripiprazole.
Hepatic impairment.
A single-dose study with oral administration of aripiprazole to subjects with varying degrees of liver cirrhosis (Child-Pugh classes A, B, and C) did not reveal a significant effect of hepatic impairment on the pharmacokinetics of aripiprazole and dehydro-aripiprazole. The AUC of aripiprazole, compared to healthy subjects, increased 31% in mild hepatic impairment, increased 8% in moderate hepatic impairment, and decreased 20% in severe hepatic impairment. None of these differences would require dose adjustment, but the study included only three patients with class C liver cirrhosis, which is insufficient to draw conclusions on their metabolic capacity.
Smoking.
Population pharmacokinetic evaluation of oral aripiprazole has revealed no evidence of clinically relevant effects from smoking on the pharmacokinetics of aripiprazole.
5.3 Preclinical Safety Data
Animal toxicology.
The toxicological profile for aripiprazole administered to experimental animals by intramuscular injection is generally similar to that seen following oral administration at comparable plasma levels of the drug. With intramuscular injection, however, injection-site tissue reactions are observed that consist of localised inflammation, swelling, scabbing and foreign-body reactions to deposited drug. These effects gradually resolved with discontinuation of dosing.
Choleliths observed in the bile of monkeys given aripiprazole orally at doses of 25 to 125 mg/kg/day for 4 to 52 weeks (1-3 times the oral MRHD of 30 mg/day based on plasma AUC and 15-76 times the oral MRHD based on mg/m2) have been attributed to precipitation of sulfate conjugates of hydroxy metabolites, which exceeded their solubility limits in bile. Human biliary concentrations of these sulfate conjugates after repeated daily administration of the oral MRHD are substantially lower (0.2-14% of their in vitro solubility limits).
Bilateral retinal degeneration was observed in albino rats given oral aripiprazole for 6 months or 2 years at exposures of 6-13 times the clinical exposure at the oral MRHD of 30 mg/day (based on plasma AUC). The exposure at the no-effect dose was 3 times that at the MRHD. A subsequent 18-month study reported this finding in albino but not pigmented rats, possibly due to lack of photoprotective ocular melanin in the albino rats, although it is unknown whether pigmentation prevented or merely delayed retinal degeneration in the pigmented rats. The clinical relevance of this finding is uncertain.
Genotoxicity.
Aripiprazole was tested for genotoxic potential in a standard range of assays for gene mutation, chromosomal damage, and DNA damage and repair. Aripiprazole was nongenotoxic in the in vitro bacterial reverse mutation assay, in vitro forward gene mutation assay in mouse lymphoma cells, in vitro bacterial DNA repair assay, and the unscheduled DNA synthesis assay in rat hepatocytes. However, aripiprazole and its minor metabolite 2,3-DCPP were clastogenic in the in vitro chromosomal aberration assay in Chinese hamster lung cells with and without metabolic activation. A positive response in 1 of 6 in vivo mouse micronucleus tests was likely due to a mechanism not relevant to humans.
Carcinogenicity.
Lifetime carcinogenicity studies were conducted in ICR mice and in Sprague-Dawley (SD) and Fischer (F344) rats. Aripiprazole was administered for 2 years in the diet at doses of 1, 3, 10, and 30 mg/kg/day to ICR mice and 1, 3, and 10 mg/kg/day to F344 rats (0.2 to 5 and 0.3 to 3 times the oral maximum recommended human dose [MRHD] of 30 mg/day based on mg/m2, respectively). SD rats were dosed orally by gavage for 2 years at 10, 20, 40, and 60 mg/kg/day (3 to 18 times the MRHD based on mg/m2). There was no evidence of tumorigenesis in male mice. In female mice, the incidences of pituitary gland adenomas and mammary gland adenocarcinomas and adenocanthomas were increased at dietary doses of 3 to 30 mg/kg/day (0.1 to 0.9 times the MRHD based on AUC and 0.5 to 5 times the MRHD based on mg/m2). In female rats, the incidence of mammary gland fibroadenomas was increased at a dietary dose of 10 mg/kg/day (< 0.1 times the MRHD based on AUC and 3 times the MRHD based on mg/m2); and the incidences of adrenocortical carcinomas and combined adrenocortical adenomas/carcinomas were increased at an oral gavage dose of 60 mg/kg/day (10 times the MRHD based on AUC and 18 times the MRHD based on mg/m2). In male rats, the incidences of benign and combined benign/malignant phaeochromocytomas were also increased at an oral gavage dose of 60 mg/kg/day (10 times the MRHD based on AUC and 18 times the MRHD based on mg/m2).
Proliferative changes in the pituitary and mammary gland of rodents have been observed following chronic administration of other antipsychotic agents and are considered prolactin mediated. Serum prolactin was not measured in the aripiprazole carcinogenicity studies. At the doses associated with mammary and/or pituitary tumours, hyperprolactinaemia was observed in female mice in a 13-week dietary study but not in female rats in 4- and 13-week dietary studies. Hyperprolactinaemia was observed in female rats after 5 and 13 weeks of oral administration at doses up to that associated with adrenocortical tumours, but serum prolactin was decreased at this dose in male rats. The relationship between tumourigenic findings with aripiprazole and prolactin is unclear and the relevance for human risk of prolactin-mediated endocrine tumours is unknown. The adrenocortical response in female rats is considered a consequence of increased adrenocortical cell proliferation secondary to chronic drug-related adrenocortical cytotoxicity; the no-effect exposure (plasma AUC) was about 7 times clinical exposure at the MRHD.6 Pharmaceutical Particulars
6.1 List of Excipients
The vial of aripiprazole 400 mg contains the following excipients: carmellose sodium, mannitol, monobasic sodium phosphate monohydrate and sodium hydroxide.
The diluent vial contains water for injections.
6.2 Incompatibilities
See Section 4.5 Interactions with Other Medicines and Other Forms of Interactions.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Therapeutic kit.
The vial containing aripiprazole (as monohydrate) lyophilised powder is stable up to the expiration date stated on the carton.
Store in the original packaging below 25°C. Do not freeze.
For single use in only one patient. Discard any unused solution.
The reconstituted suspension should be used immediately, but may be stored below 25°C for up to 4 hours. Shake the vial vigorously for at least 60 seconds to re-suspend prior to injection.
6.5 Nature and Contents of Container
Therapeutic kit.
Aripena is provided as a one-month therapeutic kit that contains:
one vial of aripiprazole 400 mg powder for suspension for injection;
one vial containing water for injections 2 mL;
one 3 mL sterile syringe with a 21 gauge needle for injection;
one 3 mL sterile syringe without a needle;
one vial adapter;
one 23 gauge, 1-inch (25 mm) sterile safety needle for injection;
one 22 gauge, 1.5-inch (38 mm) sterile safety needle for injection;
one 21 gauge, 2-inch (51 mm) sterile safety needle for injection.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Aripiprazole in Aripena is present as aripiprazole monohydrate. The chemical name of aripiprazole monohydrate is 7-[4-[4-(2,3-dichlorophenyl)-1-piperazinyl]butoxy]-3,4-dihydro-carbostyril monohydrate. The molecular formula is C23H27Cl2N3O2.H2O and its molecular weight is 466.40. Aripiprazole monohydrate is a white-to-off-white crystalline powder. Aripiprazole monohydrate is practically insoluble in water.
Chemical structure.
Aripiprazole monohydrate has the structural formula:

CAS number.
851220-85-4.7 Medicine Schedule (Poisons Standard)
S4 - Prescription Only Medicine.
Summary Table of Changes
