What is in this leaflet?
This leaflet answers some common questions about Arixtra.
It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor or pharmacist has weighed the risks of you using Arixtra against the benefits they expect it will have for you.
If you have any concerns about using this medicine, ask your doctor or pharmacist.
Please read this leaflet carefully before you start using Arixtra. You may wish to keep it to read again.
What Arixtra is used for
Arixtra is used to prevent blood clots forming in patients who are recovering from orthopaedic or abdominal surgery. Arixtra is also used to treat blood clots once they have formed. Arixtra contains the medication fondaparinux sodium, a synthetic compound which helps prevent blood clots forming in blood vessels. This type of blood clot, also called deep venous thrombosis, or DVT, can occur in patients who are confined to bed after hip or knee surgery. Arixtra is also used to treat some types of heart attacks and severe angina (a pain caused by narrowing of the arteries in the heart).
Your doctor may have prescribed Arixtra for another reason. Ask your doctor if you have any questions about why Arixtra has been prescribed for you.
Arixtra is not addictive.
This medicine is available only with a doctor's prescription.
Before you are given Arixtra
When you must not be given it
You should not be given Arixtra if you have any of the following conditions;
- You are bleeding excessively
- You have severe kidney disease
- You have acute bacterial endocarditis (an infection of the heart)
Do not use Arixtra if you have an allergy to:
- any medicine containing fondaparinux sodium
- any of the ingredients listed at the end of this leaflet
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin
Do not use Arixtra if you are pregnant or intend to become pregnant. Arixtra is not recommended for use during pregnancy, unless you and your doctor or pharmacist have discussed the risks and benefits involved.
Do not use Arixtra if you are breast-feeding or plan to breast-feed. It is not known whether Arixtra passes into human breast milk.
Do not use this medicine in children under the age of 17 years. Safety and effectiveness in children younger than 17 years have not been established.
Do not use Arixtra after the expiry date (EXP) printed on the pack. If you use this medicine after the expiry date has passed, it may not work as well as it should.
Do not use Arixtra if the packaging is torn or shows signs of tampering.
If you are not sure whether you should start using Arixtra, talk to your doctor or pharmacist.
Before you are given it
Tell your doctor or pharmacist if you have allergies to:
- any other medicines
- any other substances, such as foods, preservatives or dyes
- latex
Tell your doctor or pharmacist if you are pregnant or intend to become pregnant. Your doctor or pharmacist will discuss the possible risks and benefits of using Arixtra during pregnancy.
Tell your doctor or pharmacist if you are breast-feeding or plan to breast-feed. Your doctor or pharmacist will discuss the possible risks and benefits of using Arixtra during breast-feeding.
Tell your doctor or pharmacist if you have or have had any medical conditions, especially the following:
- a stomach ulcer
- bleeding disorders
- recent bleeding inside the head
- recent surgery on the brain, eye, or spinal column
- moderate or severe kidney disease or severe liver disease
- heparin induced thrombocytopenia (low platelet count).
Tell your doctor or pharmacist if you are elderly or have a low body weight (less than 50 kg) as you may be at increased risk of bleeding if you are given Arixtra.
It is recommended that your doctor monitor's your blood platelets at the beginning and end of your treatment with Arixtra.
Tell your doctor if you are going to have a stent inserted into your heart.
If you have not told your doctor or pharmacist about any of the above, tell them before you start using Arixtra.
Taking other medicines
Tell your doctor or pharmacist if you are taking/using any other medicines, including any that you buy without a prescription from your pharmacy, supermarket or health food shop. Some medicines and Arixtra may interfere with each other.
Your doctor or pharmacist have more information on medicines to be careful with or to avoid while using Arixtra.
How Arixtra is given
Follow all directions given to you by your doctor or pharmacist carefully.
They may differ from the information contained in this leaflet.
If you do not understand the instructions, ask your doctor or pharmacist for help.
How much is given
The usual dose of Arixtra is 2.5 mg given once a day, starting after your operation. Arixtra may be given for up to 31 days.
How it is given
Arixtra is given to you as a subcutaneous injection (an injection just under the skin). To treat some types of heart attack, the health professional only may give the first dose intravenously (into a vein).
Arixtra should NOT be given as an intramuscular injection (an injection into the muscle).
The injections can be given by a doctor or nurse, or you may be taught how to give the subcutaneous injections to yourself.
Instructions on how to use the Arixtra syringe subcutaneously are given at the end of this leaflet.
Overdose
Immediately telephone your doctor or the Poisons Information Centre (telephone Australia 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much Arixtra. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Overdose may lead to an increased risk of bleeding.
While you are using Arixtra
Things you must do
Tell any other doctors, dentists, and pharmacists who are treating you that you are using Arixtra.
If you are about to be started on any new medicine, tell your doctor, dentist or pharmacist that you are using Arixtra.
Immediately tell your doctor if you develop the following:
- pain or swelling in the legs
- chest pain or difficulty in breathing
These may be signs that a blood clot has formed.
If you have epidural or spinal anaesthesia (a pain killing injection around the spinal cord), tell your doctor or nurse immediately if you have back pain, numbness or weakness in the legs, or problems with bowel or bladder function.
Things you must not do
Do not use Arixtra to treat any other complaints unless your doctor or pharmacist tells you to. Do not give Arixtra to anyone else, even if they have the same condition as you.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are using Arixtra. Arixtra helps most people at risk of blood clots following surgery, but it may have unwanted side effects in some people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
If you get any side effects, do not stop using Arixtra without first talking to your doctor or pharmacist.
Tell your doctor or pharmacist if you notice any of the following:
- bleeding
- oozing of fluid from the wound
- unusual tiredness, weakness or drowsiness
- fever
- nausea or vomiting
- indigestion or gastritis
- low blood pressure
- fainting or vertigo
- dizziness or confusion
- difficulty sleeping
- pain or skin reactions at the injection site
- itching sensation on the skin
- urinary tract infection
- difficulty or pain when passing urine
- diarrhoea or constipation
- headache
- coughing
- swelling (oedema)
- shortness of breath
- flushing of the skin
These side effects of Arixtra are usually mild.
Tell your doctor or pharmacist immediately if you notice any of the following:
- allergic reaction. Symptoms of an allergic reaction may include:
- shortness of breath, wheezing or difficulty breathing,
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin - significant bleeding
These may be signs of serious side effects. You may need urgent medical attention. Serious side effects are rare.
The following side effects may be identified by tests performed by your doctor:
- blood disorders such as decreased or abnormal blood platelets, clotting disorders or excess bilirubin
- changes in liver function or liver enzymes
Other side effects not listed above may occur in some patients. Tell your doctor or pharmacist if you notice anything that is making you feel unwell.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
After using Arixtra
Storage
Arixtra will normally be stored in the pharmacy or on the hospital ward. The injection should be kept at room temperature (less than 25°C).
Disposal
If your doctor or pharmacist tells you to stop using Arixtra or the injections have passed their expiry date, ask your pharmacist what to do with any that are left over.
Product description
What it looks like
Arixtra comes as a pre-filled safety syringe, in pack sizes of 2 pre-filled syringes.
Ingredients
Active ingredients:
- fondaparinux sodium
Other ingredients
- sodium chloride
- water for injections
Distributor
Arixtra is supplied in Australia by;
Aspen Pharmacare Australia Pty Ltd
34-36 Chandos Street
St Leonards NSW 2065
Australia
Arixtra has the following registration No:
2.5 mg - AUST R 80279
This leaflet was updated in September 2018
Method of administration
Arixtra is administered by subcutaneous injection (an injection just under the skin). Arixtra is for single use in one patient only. Discard any residue.
The solution should be inspected visually for particles and discoloration prior to administration. If present, do not proceed with the injection.
Subcutaneous administration
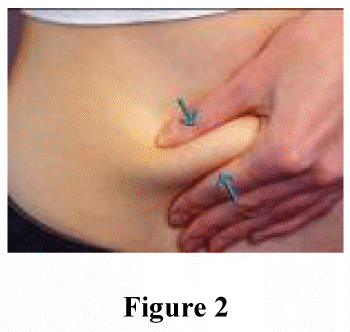
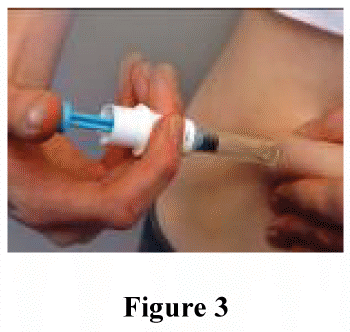
The sites of subcutaneous injection should be on the fatty tissues on the abdominal area. It is important to alternate between the left and the right sides. To avoid the loss of medicinal product when using the pre-filled syringe do not expel the air bubble from the syringe before injection. The whole length of the needle should be inserted between the thumb and forefinger (see figures 2 and 3). The skin fold should be held throughout the injection.
Arixtra is intended for use under a physician’s guidance. Patient’s may self-inject only if their physician determines that it is appropriate, and with medical follow-up as necessary. Proper training in subcutaneous injection technique should be provided. Instructions for self-administration subcutaneously are included below.
The parts of the syringe with an automatic needle protection system are:
- Needle shield
- Plunger
- Finger-grip
- Security sleeve

To use the Arixtra syringe, remove the needle shield, by first twisting it and then pulling it straight off (figure 1).
Discard the needle shield.
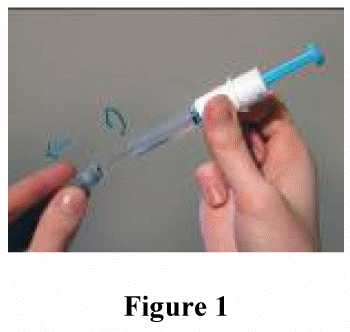
For subcutaneous administration:
Gently pinch the skin that has been cleaned to make a fold. Hold the fold between the thumb and the forefinger during the entire injection (figure 2).
Insert the full length of the needle perpendicularly (at an angle of 90°) into the skin fold (figure 3).
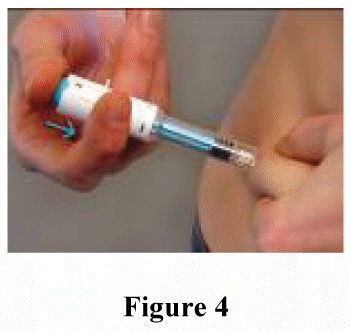
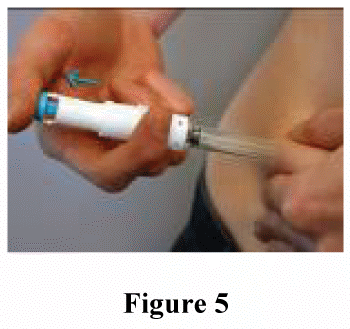
Inject ALL of the content of the syringe by pressing down on the plunger as far as it goes (figure 4), and then release it: the needle will withdraw automatically from the skin into a security sleeve and then will be locked permanently (figure 5).
Discard the used syringe in a safe manner by using a sharps disposal container. This is usually available at the local community pharmacy.
Published by MIMS March 2024
 The rates of bleeding events reported during the abdominal surgery clinical trial with Arixtra 2.5 mg injection are provided in Table 2.
The rates of bleeding events reported during the abdominal surgery clinical trial with Arixtra 2.5 mg injection are provided in Table 2. A separate analysis of major bleeding according to the time of the first injection of Arixtra after surgical closure was performed. In this analysis the incidences of major bleeding were as follows: 3.4% (9/263) if the first injection was given < 6 hours and 2.8% (32/1139) if the first injection was performed ≥ 6 hours.
A separate analysis of major bleeding according to the time of the first injection of Arixtra after surgical closure was performed. In this analysis the incidences of major bleeding were as follows: 3.4% (9/263) if the first injection was given < 6 hours and 2.8% (32/1139) if the first injection was performed ≥ 6 hours. Other adverse events that occurred during treatment with Arixtra or enoxaparin sodium in clinical trials with patients undergoing hip fracture surgery, hip replacement surgery, or knee replacement surgery and that occurred at a rate of at least 2% in either treatment group, are provided in Table 4.
Other adverse events that occurred during treatment with Arixtra or enoxaparin sodium in clinical trials with patients undergoing hip fracture surgery, hip replacement surgery, or knee replacement surgery and that occurred at a rate of at least 2% in either treatment group, are provided in Table 4.

 Other adverse reactions that occurred during treatment with Arixtra in clinical trials with patients undergoing hip fracture surgery, hip replacement surgery, knee replacement surgery or abdominal surgery and that occurred at a rate of less than 1% include the following: thrombocytopenia, thrombocythemia, abnormal platelets, coagulation disorder, pruritus, hepatic enzymes increased, hepatic function abnormal and wound secretion. Adverse reactions occurring at a rate of less than 0.1% include somnolence, vertigo, dyspnea, gastritis, bilirubinemia, fatigue, flushing and syncope.
Other adverse reactions that occurred during treatment with Arixtra in clinical trials with patients undergoing hip fracture surgery, hip replacement surgery, knee replacement surgery or abdominal surgery and that occurred at a rate of less than 1% include the following: thrombocytopenia, thrombocythemia, abnormal platelets, coagulation disorder, pruritus, hepatic enzymes increased, hepatic function abnormal and wound secretion. Adverse reactions occurring at a rate of less than 0.1% include somnolence, vertigo, dyspnea, gastritis, bilirubinemia, fatigue, flushing and syncope. The incidence of adjudicated severe haemorrhage by modified thrombolysis in myocardial infarction (TIMI) criteria was 1.1% (fondaparinux) versus 1.4% (control [UFH/placebo]) up to and including Day 9 in the Phase III STEMI study (Table 8).
The incidence of adjudicated severe haemorrhage by modified thrombolysis in myocardial infarction (TIMI) criteria was 1.1% (fondaparinux) versus 1.4% (control [UFH/placebo]) up to and including Day 9 in the Phase III STEMI study (Table 8). The risk of peri-PCI major bleeding when Arixtra was used in conjunction with UFH was low and comparable to that observed in the Phase III UA/NSTEMI study (see Section 5.1 Pharmacodynamic Properties, Clinical trials, Table 18). In the Phase III UA/NSTEMI study, the most commonly reported non-bleeding adverse events (reported in at least 1% of subjects on fondaparinux) were headache, chest pain and atrial fibrillation (Table 9).
The risk of peri-PCI major bleeding when Arixtra was used in conjunction with UFH was low and comparable to that observed in the Phase III UA/NSTEMI study (see Section 5.1 Pharmacodynamic Properties, Clinical trials, Table 18). In the Phase III UA/NSTEMI study, the most commonly reported non-bleeding adverse events (reported in at least 1% of subjects on fondaparinux) were headache, chest pain and atrial fibrillation (Table 9). In the Phase III study in STEMI patients, the most commonly reported non-bleeding adverse events (reported in at least 1% of subjects on fondaparinux) were atrial fibrillation, pyrexia, chest pain, headache, ventricular tachycardia, vomiting, and hypotension (Table 10).
In the Phase III study in STEMI patients, the most commonly reported non-bleeding adverse events (reported in at least 1% of subjects on fondaparinux) were atrial fibrillation, pyrexia, chest pain, headache, ventricular tachycardia, vomiting, and hypotension (Table 10).


 Major bleeding, all at surgical sites, was observed in 8 patients (2.4%) treated with Arixtra 2.5 mg compared to 2 (0.6%) with placebo (p = 0.063). Permanent premature treatment cessation was observed in 2 of these patients in the Arixtra group and in 1 patient in the placebo group. In each group, two of these major bleeding episodes (0.6%) led to surgical re-intervention. Death occurred in 5 randomised patients (1.5%) treated with Arixtra 2.5 mg compared to 7 (2.1%) with placebo. The 2 excess deaths with placebo were due to PE.
Major bleeding, all at surgical sites, was observed in 8 patients (2.4%) treated with Arixtra 2.5 mg compared to 2 (0.6%) with placebo (p = 0.063). Permanent premature treatment cessation was observed in 2 of these patients in the Arixtra group and in 1 patient in the placebo group. In each group, two of these major bleeding episodes (0.6%) led to surgical re-intervention. Death occurred in 5 randomised patients (1.5%) treated with Arixtra 2.5 mg compared to 7 (2.1%) with placebo. The 2 excess deaths with placebo were due to PE.

 The difference in the rate of VTE observed between patients taking fondaparinux and those taking dalteparin (4.6% and 6.1%) was not statistically significant. The PEGASUS trial indicates that fondaparinux is non-inferior to dalteparin in the prevention of DVT after abdominal surgery.
The difference in the rate of VTE observed between patients taking fondaparinux and those taking dalteparin (4.6% and 6.1%) was not statistically significant. The PEGASUS trial indicates that fondaparinux is non-inferior to dalteparin in the prevention of DVT after abdominal surgery.

 The incidences of adjudicated guiding catheter thrombus were 0.1% (1/1002) and 0.5% (5/1024), in patients randomised to "standard dose" and "low dose" UFH respectively during PCI. Four (0.3%) non-randomised patients experienced thrombus in the diagnostic catheter during coronary angiography. Of the 3,235 patients enrolled in the trial, twelve (0.37%) patients in total experienced thrombus in the arterial sheath, of these 7 cases were reported during angiography and 5 cases were reported during PCI.
The incidences of adjudicated guiding catheter thrombus were 0.1% (1/1002) and 0.5% (5/1024), in patients randomised to "standard dose" and "low dose" UFH respectively during PCI. Four (0.3%) non-randomised patients experienced thrombus in the diagnostic catheter during coronary angiography. Of the 3,235 patients enrolled in the trial, twelve (0.37%) patients in total experienced thrombus in the arterial sheath, of these 7 cases were reported during angiography and 5 cases were reported during PCI. The pharmacokinetics of fondaparinux are linear in the range of 2 to 8 mg by the subcutaneous route. At steady state, mean plasma concentration 2 hours post-dosing ranged between 0.32 and 0.47 mg/L in patients undergoing orthopaedic surgery receiving fondaparinux 2.5 mg.
The pharmacokinetics of fondaparinux are linear in the range of 2 to 8 mg by the subcutaneous route. At steady state, mean plasma concentration 2 hours post-dosing ranged between 0.32 and 0.47 mg/L in patients undergoing orthopaedic surgery receiving fondaparinux 2.5 mg. MW: 1728. C31H43N3Na10O49S8.
MW: 1728. C31H43N3Na10O49S8.