1 Name of Medicine
Ganirelix acetate.
2 Qualitative and Quantitative Composition
ARX Ganirelix 250 microgram ganirelix (as acetate)/0.5 mL solution for injection.
ARX Ganirelix contains the synthetic decapeptide ganirelix (INN) as its acetate salt, with high antagonistic activity to the naturally occurring gonadotropin releasing hormone (GnRH).
Each prefilled syringe contains 250 microgram ganirelix (as acetate) in 0.5 mL.
For the full list of excipients, see Section 6.1 List of Excipients.3 Pharmaceutical Form
Solution for injection.
ARX Ganirelix (ganirelix acetate) is presented as a sterile, ready for use, clear and colourless aqueous solution intended for subcutaneous administration.
4.1 Therapeutic Indications
For the prevention of premature luteinisation and ovulation in patients undergoing controlled ovarian stimulation, followed by oocyte pick up and assisted reproductive techniques.
4.2 Dose and Method of Administration
ARX Ganirelix should only be prescribed by a specialist experienced in the treatment of fertility.
Dosage.
ARX Ganirelix is used to prevent premature LH surges in patients undergoing COH. Controlled ovarian hyperstimulation with FSH may start at day 2 or 3 of menses. ARX Ganirelix (0.25 mg) should be injected subcutaneously once daily, starting from day 5 or day 6 of FSH administration depending on the level of ovarian response. ARX Ganirelix should not be mixed with FSH but both preparations should be administered approximately at the same time. Daily treatment with ARX Ganirelix should be continued up to the day that sufficient follicles of adequate size are present. Final maturation of follicles can be induced by administering hCG. Because of the half-life of ganirelix, the time between two ARX Ganirelix injections and between the last ARX Ganirelix injection and the hCG injection should not exceed 30 hrs, as otherwise a premature LH surge may occur.
Method of administration.
Inspect the solution before use. It must only be used if it is clear and without particulate matter.
ARX Ganirelix should be administered subcutaneously. The injection site should be varied to prevent lipoatrophy. The patient or her partner may perform the injections of ARX Ganirelix themselves, provided that they are adequately instructed and have access to expert advice. Air bubble(s) may be seen in the pre-filled syringe. This is expected, and removal of the air bubble(s) is not needed.
Product is for single use in one patient only. Discard any residue.4.3 Contraindications
Hypersensitivity to the active substance or to any of the components, see Section 6.1 List of Excipients.
Hypersensitivity to GnRH or any other GnRH analogue.
Pregnancy or lactation.
Moderate to severe renal impairment and hepatic impairment.
4.4 Special Warnings and Precautions for Use
Special care should be taken in women with signs and symptoms of active allergic conditions. Cases of hypersensitivity reactions (both generalised and local), have been reported with ARX Ganirelix, as early as with the first dose, during post-marketing surveillance. These events have included anaphylaxis (including anaphylactic shock), angioedema, and urticaria. (See Section 4.8 Adverse Effects (Undesirable Effects)). If a hypersensitivity reaction is suspected, ARX Ganirelix should be discontinued and appropriate treatment administered. Use of ARX Ganirelix in patients with active allergic symptoms has not been investigated. Administration of ARX Ganirelix is not advised to patients with currently severe allergic symptoms. Patients should be advised to contact the attending physician before administering the next injection in case a general or an extensive local allergic reaction occurs.
Ovarian hyperstimulation syndrome (OHSS) may occur during or following ovarian stimulation. OHSS must be considered an intrinsic risk of gonadotrophin stimulation. OHSS should be treated symptomatically, e.g. with rest, intravenous infusion of electrolyte solutions or colloids and heparin.
Since infertile women undergoing assisted reproduction, and particularly IVF, often have tubal abnormalities, the incidence of ectopic pregnancies might be increased. Early ultrasound confirmation that a pregnancy is intrauterine is therefore important.
The safety and efficacy of ARX Ganirelix have not been established in women weighing less than 50 kg or more than 90 kg.
Congenital abnormalities.
The incidence of congenital malformations after Assisted Reproductive Technologies (ART) may be slightly higher than after spontaneous conceptions. This slightly higher incidence is thought to be related amongst other factors, to differences in parental characteristics (e.g. maternal age, sperm characteristics) and by the higher incidence of multiple gestations after ART. In clinical trials investigating more than 1000 newborns it has been demonstrated that the incidence of congenital malformation in children born after Controlled Ovarian Hyperstimulation (COH) treatment using ARX Ganirelix is comparable with that reported after COH treatment using a GnRH agonist.
Use in hepatic impairment.
See Section 4.3 Contraindications.
Use in renal impairment.
See Section 4.3 Contraindications.
Use in the elderly.
No data available.
Paediatric use.
There is no relevant use of ARX Ganirelix in the paediatric population.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
In clinical studies, interactions of ARX Ganirelix with other medicines have not been investigated. Therefore, interactions with commonly used medicinal products cannot be excluded.
In the absence of incompatibility studies, this medicinal product must not be mixed with other medicinal products.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Ganirelix treatment of female rats resulted in reversible impairment of mating and fertility at a subcutaneous dose of 2.5 microgram/kg/day, and reversible cessation of mating was seen in males treated with a subcutaneous dose of 0.1 mg/kg/day.
(Category D)
ARX Ganirelix is not intended to be used during pregnancy (see Section 4.3 Contraindications). No clinical data on exposed pregnancies are available.
Studies in animals have indicated that ganirelix increased the incidence of total fetal resorptions when administered to pregnant rats and rabbits during the period of organogenesis, at respective doses of 10 microgram/kg/day and 30 microgram/kg/day (approximately 0.4 and 3 times the human dose, based on body surface area). The effects on fetal resorption are logical consequences of the alteration in hormonal levels brought about by the antigonadotrophic properties of ARX Ganirelix and could result in fetal loss in humans. There was no increase in fetal abnormalities. No treatment related changes in fertility, physical or behavioural characteristics were observed in the offspring of female rats treated with ganirelix during pregnancy and lactation.
ARX Ganirelix should not be used by lactating women (see Section 4.3 Contraindications). It is not known whether ganirelix is excreted into animal or human breast milk. Subcutaneous ganirelix doses of 2.5 microgram/kg/day given to lactating rats did not result in impairment of postnatal development of the offspring.4.7 Effects on Ability to Drive and Use Machines
The effects of this medicine on a person's ability to drive and use machines were not assessed as part of its registration.
4.8 Adverse Effects (Undesirable Effects)
General disorders and administrative site conditions.
Ganirelix acetate may cause a skin reaction (in 10% - 15% of the patients moderate or severe redness with or without swelling was reported) at the site of injection, which normally disappears within 4 hours after administration. Malaise was reported in 0.3% of patients.
Immune system disorders.
Very rarely, post-marketing cases of hypersensitivity reactions (including rash, facial swelling, dyspnea, anaphylaxis (including anaphylactic shock), angioedema, and urticaria) have been reported, as early as with the first dose, among patients administered ganirelix acetate.
Nervous system disorders.
Headache (0.4%).
Gastrointestinal disorders.
Nausea (0.5%).
Other reported adverse events are rather related to the controlled ovarian hyperstimulation treatment for assisted reproduction technique (ART) than to ganirelix acetate (e.g. pelvic pain, abdominal distension, ovarian hyperstimulation syndrome (OHSS), ectopic pregnancy and spontaneous abortion).
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at http://www.tga.gov.au/reporting-problems and contact Arrotex Medical Information Enquiries/Adverse Drug Reaction Reporting on 1800 195 055.4.9 Overdose
Overdosage in humans may result in a prolonged duration of action. In case of overdose, ganirelix acetate treatment should be (temporarily) discontinued.
No data on acute toxicity of ganirelix acetate in humans are available. Clinical studies with subcutaneous administration of ganirelix acetate at single doses up to 12 mg did not show systemic side-effects. Clinical signs of systemic toxicity including collapse, laboured respiration and inactivity in rats or facial flushing in monkeys were observed after intravenous administration of ganirelix at 2.0 and 3.0 mg/kg, respectively. Blood pressure was also reduced by about 50% in rats treated with an intravenous ganirelix dose of 0.9 mg/kg.
For information on the management of overdose, contact the Poisons Information Centre on 131126 (Australia).
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: anti-gonadotrophin releasing hormone; ATC code: H01CC01.
Mechanism of action.
Ganirelix acetate is a GnRH antagonist, which modulates the hypothalamic-pituitary-gonadal axis by competitively binding to the GnRH receptors in the pituitary gland. As a result, a rapid, reversible suppression of endogenous gonadotropins occurs, without initial stimulation as induced by GnRH agonists. The inhibitory effect of ganirelix acetate on the release of LH is more pronounced than on FSH. When LH has started to rise prior to the first administration of ganirelix acetate, a premature LH surge can still be prevented.
Important features of the GnRH-antagonist regimen:
within a few hours suppression of gonadotropin release due to GnRH receptor blockade;
ganirelix acetate treatment is restricted to those days when a premature LH surge may occur. Therefore, the overall duration of treatment is only several days;
less suppression of endogenous FSH and therefore less FSH required;
recovery of pituitary functioning within two days following discontinuation of the treatment;
the competitive mode of action of ganirelix acetate may allow the administration of a GnRH agonist instead of hCG to trigger ovulation, which is especially relevant for patients at risk of developing OHSS;
generally, estradiol levels are lower (though remaining above natural cycle levels) compared to the relatively high levels in the agonist regimen.
Clinical trials.
The efficacy of ganirelix acetate injection was established in three clinical studies. In these studies, the administration of exogenous recombinant FSH [Puregon (follitropin beta for injection)] was initiated on the morning of Day 2 or 3 of a natural menstrual cycle. Ganirelix acetate was administered on the morning of Day 7 or 8 (Day 6 of recombinant FSH administration). The dose of recombinant FSH administered was adjusted according to individual responses starting on the day of initiation of ganirelix acetate. Both recombinant FSH and ganirelix acetate were continued daily until at least three follicles were 17 mm or greater in diameter at which time hCG [Pregnyl (chorionic gonadotropin for injection)] was administered. Following hCG administration, ganirelix acetate and recombinant FSH administration were discontinued. Oocyte retrieval, followed by in vitro fertilisation (IVF) or intracytoplasmic sperm injection (ICSI), was subsequently performed.
In a multicenter, double blind, randomised, dose finding study (protocol 38602), ganirelix acetate doses ranging from 62.5 microgram to 2,000 microgram and recombinant FSH were administered to 332 patients undergoing COH for IVF. The results of the selected dose (250 microgram) are summarized in Table 1.
Two multicentre, open-label, randomised studies (protocol 38607 and 103-001) were conducted in which follicular phase treatment with ganirelix acetate 250 microgram was studied using a GnRH agonist as a reference treatment. A total of 661 subjects were treated with ganirelix acetate by subcutaneous injection once daily starting on Day 6 of recombinant FSH treatment. Recombinant FSH was maintained at 150 IU or 225 IU in the 38607 and 103-001 study, respectively for the first 5 days of ovarian stimulation and was then adjusted by the investigator on the sixth day of gonadotropin use according to individual responses. The results for the ganirelix acetate arm are summarized in Table 1.
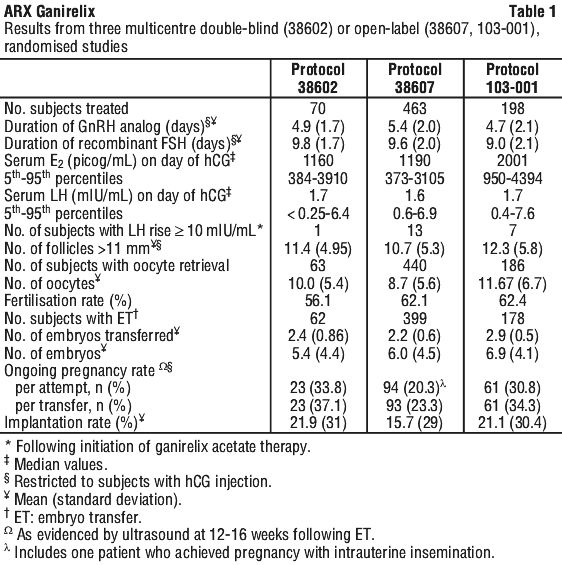 Some centres were limited to the transfer of ≤ 2 embryos based on local practice standards.
Some centres were limited to the transfer of ≤ 2 embryos based on local practice standards.
In subjects administered ganirelix acetate 250 microgram, a premature LH surge prior to hCG administration, (LH rise ≥ 10 mIU/mL with a significant rise in serum progesterone > 2 nanogram/mL, or a significant decline in serum estradiol) occurred in less than 1% of subjects.
In case of high ovarian response, as assessed by the number and size of growing follicles and/or the amount of circulating estradiol, either as a result of a high FSH exposure in the early follicular phase or as a result of high ovarian responsiveness, premature LH rises may occur earlier than day 6 of stimulation. Initiation of ganirelix acetate treatment on day 5 can effectively prevent these premature LH rises without compromising the clinical outcome.
5.2 Pharmacokinetic Properties
Absorption.
After a single subcutaneous administration of 250 microgram, serum levels of ganirelix rise rapidly and reach peak levels (Cmax) of approximately 15 nanogram/mL within 1 to 2 hrs (tmax). The bioavailability of ganirelix acetate following subcutaneous administration is approximately 91%.
Pharmacokinetic parameters after multiple subcutaneous dosing of ganirelix acetate (once daily injection) were similar to those after a single subcutaneous dose. After repeated dosing at 0.25 mg/day, steady-state levels of approximately 0.6 nanogram/mL were reached within 2 to 3 days.
Distribution.
The mean (SD) volume of distribution of ganirelix acetate in healthy females following intravenous administration of a single 250 microgram dose is approximately 44 (± 11) liters. In vitro protein binding to human plasma is approximately 82%.
Metabolism.
The major circulating component in plasma is ganirelix. Ganirelix is also the main compound found in urine, and faeces only contained metabolites.
Excretion.
After a single subcutaneous administration of 250 microgram, the elimination half-life (t1/2) is approximately 13 hrs and clearance is approximately 2.4 L/h.
In a radiolabelled study (n = 3), ganirelix acetate was excreted via faeces (approximately 75%) and urine (approximately 22%).
5.3 Preclinical Safety Data
Genotoxicity.
Ganirelix showed no evidence of genotoxicity in assays for gene mutation in bacterial or mammalian cells. Ganirelix was not clastogenic in tests for chromosomal damage in Chinese hamster ovary cells in vitro and for micronucleus formation in mice in vivo.
Carcinogenicity.
Long-term carcinogenicity studies with ganirelix have not been carried out.6 Pharmaceutical Particulars
6.1 List of Excipients
The ARX Ganirelix solution for injection also contains glacial acetic acid, mannitol and water for injections. The pH may have been adjusted with sodium hydroxide and/or glacial acetic acid.
6.2 Incompatibilities
See Section 4.2 Dose and Method of Administration; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store at room temperature below 30°C, in the original package. Do not freeze as the syringe may break. Protect from light.
6.5 Nature and Contents of Container
ARX Ganirelix is supplied in disposable prefilled syringes (siliconised Type I glass), containing 250 microgram ganirelix/0.5 mL. Each prefilled syringe is affixed with a needle closed by a needle shield (see Section 4.4 Special Warnings and Precautions for Use).
Boxes of ARX Ganirelix contain 1 or 5 prefilled syringes.
6.6 Special Precautions for Disposal
This product is for single use only. Discard any remaining contents.
Any unused product or waste material should be disposed of in accordance with local requirements.
6.7 Physicochemical Properties
The amino acids at positions 1, 2, 3, 6, 8 and 10 of the natural GnRH decapeptide have been substituted resulting in Ac-D-Nal-D-4-CI-Phe-D-Pal-L-Ser-L-Tyr-diEt-hArg-L-Leu-diEt- hArg-L- Pro-D-Ala-NH2 with a molecular weight of 1570.4 (anhydrous free base).
Molecular formula: C80H113N18O13Cl as the free base.
Chemical structure.

CAS number.
The CAS Registry numbers are 124904-93-4 (free base); 129311-55-3 (acetate).7 Medicine Schedule (Poisons Standard)
Prescription Only Medicine (Schedule 4).
Summary Table of Changes
