1 Name of Medicine
Oxycodone hydrochloride and naloxone hydrochloride.
2 Qualitative and Quantitative Composition
ARX-Oxycodone/Naloxone 2.5/1.25 modified release tablets contain oxycodone hydrochloride 2.5 mg/naloxone hydrochloride 1.25 mg.
ARX-Oxycodone/Naloxone 5/2.5 modified release tablets contain oxycodone hydrochloride 5 mg/naloxone hydrochloride 2.5 mg.
ARX-Oxycodone/Naloxone 10/5 modified release tablets contain oxycodone hydrochloride 10 mg/naloxone hydrochloride 5 mg.
ARX-Oxycodone/Naloxone 15/7.5 modified release tablets contain oxycodone hydrochloride 15 mg/naloxone hydrochloride 7.5 mg.
ARX-Oxycodone/Naloxone 20/10 modified release tablets contain oxycodone hydrochloride 20 mg/naloxone hydrochloride 10 mg.
ARX-Oxycodone/Naloxone 30/15 modified release tablets contain oxycodone hydrochloride 30 mg/naloxone hydrochloride 15 mg.
ARX-Oxycodone/Naloxone 40/20 modified release tablets contain oxycodone hydrochloride 40 mg/naloxone hydrochloride 20 mg.
ARX-Oxycodone/Naloxone 60/30 modified release tablets contain oxycodone hydrochloride 60 mg/naloxone hydrochloride 30 mg.
ARX-Oxycodone/Naloxone 80/40 modified release tablets contain oxycodone hydrochloride 80 mg/naloxone hydrochloride 40 mg.
The inactive ingredients in the ARX-Oxycodone/Naloxone modified release tablet core are ethylcellulose, stearyl alcohol, purified talc and magnesium stearate.
ARX-Oxycodone/Naloxone modified release tablets 2.5/1.25 mg, 5/2.5 mg and 15/7.5 mg also contain hyprolose.
ARX-Oxycodone/Naloxone modified release tablets 10/5 mg, 20/10 mg, 30/15 mg, 40/20 mg, 60/30 mg and 80/40 mg also contain povidone.
The tablets are coated with polyvinyl alcohol, titanium dioxide, macrogol 3350 and purified talc. The tablet's film coating also contains:
2.5/1.25 mg.
Opadry II complete film coating system 85F220026 yellow (ARTG PI No: 108764).
5/2.5 mg.
Brilliant blue FCF aluminium lake (ARTG No: 104464).
15/7.5 mg.
Opadry II complete film coating system 85F275005 grey (ARTG PI No: 108768).
20/10 mg.
Iron oxide red (ARTG No: 93285).
30/15 mg.
Opadry II complete film coating system 85F265010 brown (ARTG PI No: 108770).
40/20 mg.
Iron oxide yellow (ARTG No: 93287).
60/30 mg.
Opadry II complete film coating system 85F250018 red (ARTG PI No: 110098).
80/40 mg.
Opadry II complete film coating system 85F210069 brown (ARTG PI No: 110093).
Excipients with known effect.
Contains sugars as lactose and sulfites.
May contain presence of sulfites from the manufacturing process.3 Pharmaceutical Form
ARX-Oxycodone/Naloxone modified release tablets are available* as round or capsule shaped, unscored film-coated modified release tablets in blister pack sizes of 20, 28 and 60 modified release tablets as follows:
2.5/1.25 mg round, light yellow tablets without embossing;
5/2.5 mg capsule shaped, blue tablets, marked "OXN" on one side and "5" on the other;
10/5 mg capsule shaped, white tablets, marked "OXN" on one side and "10" on the other;
15/7.5 mg capsule shaped, grey tablets, marked "OXN" on one side and "15" on the other;
20/10 mg capsule shaped, pink tablets, marked "OXN" on one side and "20" on the other;
30/15 mg capsule shaped, brown tablets, marked "OXN" on one side and "30" on the other;
40/20 mg capsule shaped, yellow tablets, marked "OXN" on one side and "40" on the other;
60/30 mg capsule shaped, red tablets, marked with "OXN" on one side and "60" on the other;
80/40 mg capsule shaped, brown tablets, marked with "OXN" on one side and "80" on the other.
*Not all strengths and pack sizes are currently marketed in Australia.
4.1 Therapeutic Indications
ARX-Oxycodone/Naloxone modified release tablet is indicated for the management of severe pain where:
other treatment options have failed, are contraindicated, not tolerated or are otherwise inappropriate to provide sufficient management of pain; and
the pain is opioid-responsive; and
requires daily, continuous, long term treatment.
ARX-Oxycodone/Naloxone modified release tablet is not indicated for use in chronic non-cancer pain other than in exceptional circumstances.
ARX-Oxycodone/Naloxone modified release tablet is not indicated as an as-needed (PRN) analgesia.
The naloxone component in a fixed combination with oxycodone is indicated for the therapy and/or prophylaxis of opioid-induced constipation.
ARX-Oxycodone/Naloxone is indicated as a second line symptomatic treatment of patients with severe to very severe idiopathic restless legs syndrome after failure of dopaminergic therapy.
4.2 Dose and Method of Administration
ARX-Oxycodone/Naloxone modified release tablets are to be swallowed whole and are not to be broken, chewed or crushed. Taking broken, chewed or crushed tablets could lead to the rapid release and absorption of a potentially toxic dose of oxycodone that could be fatal.
ARX-Oxycodone/Naloxone modified release tablets 60/30 mg and 80/40 mg, should only be used in opioid-tolerant patients. In patients not previously exposed to opioids (opioid naïve), these tablet strengths may cause fatal respiratory depression.
Before initiating treatment with ARX-Oxycodone/Naloxone, a treatment strategy including treatment duration and treatment goals, and a plan for end of the treatment, should be agreed together with the patient, in accordance with pain management guidelines. During treatment, there should be frequent contact between the physician and the patient to evaluate the need for continued treatment, consider discontinuation and to adjust dosages if needed. When a patient no longer requires therapy with oxycodone, it may be advisable to taper the dose gradually to prevent symptoms of withdrawal. In absence of adequate pain control, the possibility of hyperalgesia, tolerance and progression of underlying disease should be considered (see Section 4.4 Special Warnings and Precautions for Use).
ARX-Oxycodone/Naloxone should not be used longer than necessary.
Analgesia.
Adults and elderly.
Prior to initiation and titration of doses, see Section 4.4 Special Warnings and Precautions for Use for information on special risk groups. The usual starting dose for opioid-naïve patients or patients presenting with severe pain uncontrolled by weaker opioids is 10/5 mg 12-hourly. Two lower strengths (2.5/1.25 mg and 5/2.5 mg) are available to facilitate dose titration when initiating opioid therapy, and individual dose adjustment. One ARX-Oxycodone/Naloxone modified release tablet 5/2.5 mg taken 12-hourly, or one ARX-Oxycodone/Naloxone modified release tablet 2.5/1.25 mg taken 12-hourly are suitable for patients with mild hepatic impairment or for patients with renal impairment. The dose should then be cautiously titrated, as frequently as every 1-2 days if necessary, to achieve pain relief.
Patients already being treated with opioids may be started on higher doses of ARX-Oxycodone/Naloxone tablets, depending upon their previous opioid exposure.
Patients transferring from other opioid formulations.
Patients receiving other oral oxycodone formulations may be transferred to ARX-Oxycodone/Naloxone tablets at the same total daily dosage, equally divided into two 12-hourly ARX-Oxycodone/Naloxone tablets doses.
For patients who are receiving an alternative opioid, the "oral oxycodone equivalent" of the analgesic presently being used should be determined. Having determined the total daily dosage of the present analgesic, the following equivalence table (Table 1) can be used to calculate the approximate daily oral oxycodone dosage that should provide equivalent analgesia. The total daily oral oxycodone dosage should then be equally divided into two 12-hourly ARX-Oxycodone/Naloxone tablet doses.
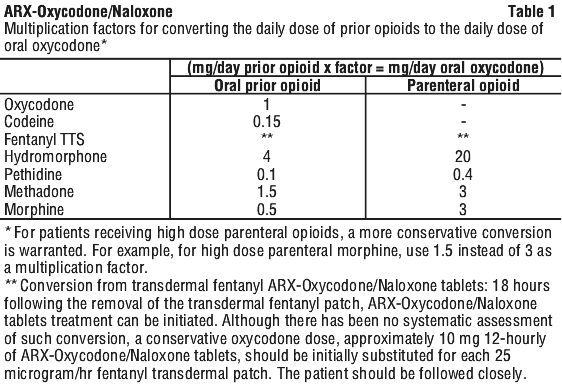 It is emphasised that this is a guide to the required dose of ARX-Oxycodone/Naloxone modified release tablets only. Inter-patient variability requires that each patient is carefully titrated to the appropriate dose. The correct dosage for any individual patient is the minimum dose that controls the pain, provides functional improvement and is well tolerated, for a full 12 hours. Patients should be titrated to pain relief and functional improvement unless unmanageable adverse drug reactions prevent this.
It is emphasised that this is a guide to the required dose of ARX-Oxycodone/Naloxone modified release tablets only. Inter-patient variability requires that each patient is carefully titrated to the appropriate dose. The correct dosage for any individual patient is the minimum dose that controls the pain, provides functional improvement and is well tolerated, for a full 12 hours. Patients should be titrated to pain relief and functional improvement unless unmanageable adverse drug reactions prevent this.
Some patients taking ARX-Oxycodone/Naloxone tablets may require "rescue" medication for breakthrough pain. ARX-Oxycodone/Naloxone tablets are a prolonged release formulation and are not intended to treat breakthrough pain. Should breakthrough pain treatment be necessary, it is generally recommended that a single dose of rescue medication should be approximately 1/6 to 1/12 of the equivalent daily dose of oxycodone hydrochloride. The need for more than two doses of "rescue" medication per day is usually an indication for the patient to be re-assessed and, if appropriate, the dosage of ARX-Oxycodone/Naloxone tablets increased.
The maximum recommended daily dose of ARX-Oxycodone/Naloxone tablets is 160/80 mg (12-hourly ARX-Oxycodone/Naloxone tablets 80/40 mg). Patients requiring higher dosages should be administered supplemental, single entity controlled release oxycodone at the same time intervals. In the case of supplemental oxycodone dosing, the beneficial effect of naloxone on bowel function may be impaired. After complete discontinuation of ARX-Oxycodone/Naloxone modified release tablets and a subsequent switch to another opioid, a worsening of bowel function can be expected.
Non-cancer pain.
Daily doses of up to 40/20 mg ARX-Oxycodone/Naloxone tablets are usually sufficient for the treatment of non-cancer pain, but higher doses may be required. ARX-Oxycodone/Naloxone tablets should not be prescribed and taken by the patient for longer than absolutely necessary. If long-term treatment is anticipated given the nature and severity of the illness, careful and regular assessment and monitoring is required to establish the clinical need for ongoing treatment with an opioid therapy.
Missed dose.
If the patient forgets to take a scheduled dose of ARX-Oxycodone/Naloxone tablets, they should be instructed not to take their next dose unless it is more than 8 hours before their next regularly scheduled dose. If so, they should be instructed to take the missed dose and return to their original dosing schedule, every 12 hours. Patients should be advised not to take extra tablets or a double dose to make up for a missed dose.
Restless legs syndrome.
ARX-Oxycodone/Naloxone tablets are indicated for patients suffering from RLS for at least 6 months. RLS symptoms should be severe and present daily and during daytime (≥ 4 days/week). ARX-Oxycodone/Naloxone tablets should be used after failure of previous dopaminergic treatment. Dopaminergic treatment failure is defined as inadequate initial response, a response that has become inadequate with time, occurrence of augmentation or unacceptable tolerability despite adequate doses. Previous treatment with at least one dopaminergic medicinal product should have lasted in general 4 weeks. A shorter period might be acceptable in case of unacceptable tolerability with dopaminergic therapy. There is no clinical experience in administration of ARX-Oxycodone/Naloxone modified release tablets with dopaminergic medication for the treatment of RLS. The combination was not assessed in the clinical trial OXN3502 in RLS patients.
Treatment should be under the supervision of a clinician with experience in the management of RLS.
At least every three months during therapy patients should be clinically evaluated and treatment should only be continued if ARX-Oxycodone/Naloxone tablets are considered effective and the benefit is considered to outweigh adverse effects and potential harms in individual patients.
Prior to continuation of RLS treatment beyond 1 year a discharge regimen by gradually reducing the dose over a period of approximately one week should be considered to establish if continued treatment with ARX-Oxycodone/Naloxone tablets is indicated.
Adults and elderly.
The usual starting dose for an opioid naïve patient is 5 mg/2.5 mg of oxycodone hydrochloride/naloxone hydrochloride 12 hourly. Titration on a weekly basis is recommended in case higher doses are required.
Analgesia/ restless legs syndrome.
Pain in the majority of patients is well managed by the symmetric administration (identical morning and evening doses) of ARX-Oxycodone/Naloxone modified release tablets at the established, stable, 12-hourly fixed dosage schedule. However, some patients may benefit from an asymmetric dosing schedule (higher dose in the morning or evening) tailored to their analgesic needs, depending on the nature of their variable, diurnal pain severity. In these patients, the lowest total daily analgesic dose that provides adequate pain relief should always still be prescribed.
Use in patients under 18 years of age.
Not recommended for use in children.
Patients with impaired hepatic function.
Caution must be exercised when administering ARX-Oxycodone/Naloxone tablets to patients with mild hepatic and renal impairment (see Section 4.4 Special Warnings and Precautions for Use, Use in renal impairment and Use in hepatic impairment).
In patients with moderate and severe hepatic impairment ARX-Oxycodone/Naloxone tablets is contraindicated (see Section 4.3 Contraindications).4.3 Contraindications
Hypersensitivity to opioids, naloxone and any of the excipients or any situation where opioids are contraindicated; moderate to severe hepatic impairment; elevated carbon dioxide levels in the blood, severe respiratory disease, acute respiratory disease and respiratory depression; cor pulmonale; cardiac arrhythmias; uncontrolled bronchial asthma; severe chronic obstructive pulmonary disease; non-opioid induced paralytic ileus; pregnancy; lactation; severe CNS depression; increased cerebrospinal or intracranial pressure; brain tumour or head injury (due to the risk of increased intracranial pressure); uncontrolled convulsive disorders; suspected surgical abdomen; delayed gastric emptying; alcoholism; delirium tremens; concurrent administration of monoamine oxidase inhibitors (MAOIs) and for 2 weeks after their cessation. History of opioid abuse for restless legs syndrome (RLS).
4.4 Special Warnings and Precautions for Use
Hazardous and harmful use.
ARX-Oxycodone/Naloxone modified release tablets contain the opioid oxycodone hydrochloride and is a potential drug of abuse, misuse and addiction. Addiction can occur in patients appropriately prescribed ARX-Oxycodone/Naloxone modified release tablets at recommended doses.
The risk of addiction is increased in patients with a personal or family history of substance abuse (including alcohol and prescription and illicit drugs) or mental illness. The risk also increases the longer the drug is used and with higher doses. Patients should be assessed for their risks for opioid abuse or addiction prior to being prescribed ARX-Oxycodone/Naloxone modified release tablets.
All patients receiving opioids should be routinely monitored for signs of misuse and abuse. Opioids are sought by people with addiction and may be subject to diversion. Strategies to reduce these risks include prescribing the drug in the smallest appropriate quantity and advising the patient on the safe storage and proper disposal of any unused drug (see Section 6.4 Special Precautions for Storage; Section 6.6 Special Precautions for Disposal). Caution patients that abuse of oral or transdermal forms of opioids by parenteral administration can result in serious adverse events, which may be fatal.
Tolerance and physical and/or psychological dependence may develop upon repeated administration of opioids such as oxycodone. Repeated use of ARX-Oxycodone/Naloxone can lead to opioid use disorder (OUD). A higher dose and longer duration of opioid treatment can increase the risk of developing OUD. Abuse or intentional misuse of ARX-Oxycodone/Naloxone may result in overdose and/or death. The risk of developing OUD is increased in patients with a personal or a family history (parents or siblings) of substance use disorders (including alcohol use disorder), in current tobacco users or in patients with a personal history of other mental health disorders (e.g. major depression, anxiety and personality disorders).
Before initiating treatment with ARX-Oxycodone/Naloxone and during the treatment, treatment goals and a discontinuation plan should be agreed with the patient (see Section 4.2 Dose and Method of Administration). Before and during treatment the patient should also be informed about the risks and signs of OUD. If these signs occur, patients should be advised to contact their physician.
Patients will require monitoring for signs of drug-seeking behaviour (e.g. too early requests for refills). This includes the review of concomitant opioids and psycho-active drugs (like benzodiazepines). For patients with signs and symptoms of OUD, consultation with an addiction specialist should be considered.
If abused parenterally or intranasally by individuals dependent on opioid agonists, such as heroin, morphine or methadone, ARX-Oxycodone/Naloxone modified release tablets are expected to produce marked withdrawal symptoms due to the opioid receptor antagonist characteristics of naloxone, or to intensify already present withdrawal symptoms.
Parenteral injection of the tablet constituents, especially talc, can be expected to result in local tissue necrosis, pulmonary granulomas and serious adverse reactions which may be fatal.
Patients should be advised not to share ARX-Oxycodone/Naloxone modified release tablets with anyone else.
Respiratory depression.
Serious, life-threatening or fatal respiratory depression can occur with the use of opioids even when used as recommended. It can occur at any time during the use of ARX-Oxycodone/Naloxone modified release tablets, but the risk is greatest during initiation of therapy or following an increase in dose. Patients should be monitored closely for respiratory depression at these times.
The risk of life-threatening respiratory depression is also higher in elderly, frail, or debilitated patients in patients with renal and hepatic impairment and in patients with existing impairment of respiratory function (e.g. chronic obstructive pulmonary disease; asthma). Opioids should be used with caution and with close monitoring in these patients (see Section 4.2 Dose and Method of Administration). The use of opioids is contraindicated in patients with severe respiratory disease, acute respiratory disease and respiratory depression (see Section 4.3 Contraindications).
Respiratory depression occurs most frequently in overdose situations in those suffering from conditions accompanied by hypoxia when even moderate doses may dangerously decrease respiration.
The risk of respiratory depression is greater with the use of high doses of opioids, especially high potency and modified release formulations, and in opioid naïve patients. Initiation of opioid treatment should be at the lower end of the dosage recommendations with careful titration of doses to achieve effective pain relief. Careful calculation of equianalgesic doses is required when changing opioids or switching from immediate release to modified release formulations, together with consideration of pharmacological differences between opioids. Consider starting the new opioid at a reduced dose to account for individual variation in response. (see Section 4.2 Dose and Method of Administration).
Sleep related breathing disorders.
Opioids can cause sleep-related breathing disorders including central sleep apnoea (CSA) and sleep-related hypoxemia. Opioid use can increase the risk of CSA in a dose-dependent fashion. In patients who present with CSA, consider decreasing the total opioid dosage. Opioids may also cause worsening of pre-existing sleep apnoea (see Section 4.8 Adverse Effects (Undesirable Effects)).
ARX-Oxycodone/Naloxone modified release tablets should be used with extreme caution in patients with sleep apnoea and patients with a substantially decreased respiratory reserve. Severe pain antagonises the respiratory depressant effects of opioids. However, should pain suddenly subside, these effects may rapidly become manifest.
Risks from concomitant use of benzodiazepines or other CNS depressants, including alcohol.
Concomitant use of opioids and benzodiazepines or other CNS depressants, including alcohol, may result in sedation, respiratory depression, coma and death. Because of these risks, concomitant prescribing of ARX-Oxycodone/Naloxone modified release tablets with CNS depressant medicines, such as other opioid analgesics benzodiazepines, gabapentinoids, cannabis, sedatives, hypnotics, tricyclic antidepressants, antipsychotics, antihistamines, centrally-active anti-emetics and other CNS depressants, should be reserved for patients for whom other treatment options are not possible. If a decision is made to prescribe ARX-Oxycodone/Naloxone modified release tablets concomitantly with any of the medicines, the lowest effective dose should be used, and the duration of treatment should be as short as possible.
Patients should be followed closely for signs and symptoms of respiratory depression and sedation. Patients and their caregivers should be made aware of these symptoms. Patients and their caregivers should also be informed of the potential harms of consuming alcohol while taking ARX-Oxycodone/Naloxone modified release tablets.
Use of opioids in chronic (long-term) non-cancer pain (CNCP).
Opioid analgesics have an established role in the treatment of acute pain, cancer pain and palliative and end-of-life care. Current evidence does not generally support opioid analgesics in improving pain and function for most patients with chronic non-cancer pain. The development of tolerance and physical dependence and risks of adverse effects, including hazardous and harmful use, increase with the length of time a patient takes an opioid. The use of opioids for long-term treatment of CNCP is not recommended.
The use of an opioid to treat CNCP should only be considered after maximised non-pharmacological and non-opioid treatments have been tried and found ineffective, not tolerated or otherwise inadequate to provide sufficient management of pain. Opioids should only be prescribed as a component of comprehensive multidisciplinary and multimodal pain management.
Opioid therapy for CNCP should be initiated as a trial in accordance with clinical guidelines and after a comprehensive biopsychosocial assessment has established a cause for the pain and the appropriateness of opioid therapy for the patient (see Section 4.4 Special Warnings and Precautions for Use, Hazardous and harmful use, above). The expected outcome of therapy (pain reduction rather than complete abolition of pain, improved function and quality of life) should be discussed with the patient before commencing opioid treatment, with agreement to discontinue treatment if these objectives are not met.
Owing to the varied response to opioids between individuals, it is recommended that all patients be started at the lowest appropriate dose and titrated to achieve an adequate level of analgesia and functional improvement with minimum adverse reactions. Immediate-release products should not be used to treat chronic pain but may be used for a short period in opioid-naïve patients to develop a level of tolerance before switching to a modified-release formulation. Careful and regular assessment and monitoring is required to establish the clinical need for ongoing treatment. Discontinue opioid therapy if there is no improvement of pain and/or function during the trial period or if there is any evidence of misuse or abuse. Treatment should only continue if the trial has demonstrated that the pain is opioid responsive and there has been functional improvement. The patient's condition should be reviewed regularly, and the dose tapered off slowly if opioid treatment is no longer appropriate (see Section 4.4 Special Warnings and Precautions for Use, Ceasing opioids).
One doctor only should be responsible for the prescription and monitoring of the patient's opioid use. Prescribers should consult appropriate clinical guidelines on the use of opioid analgesics in such patients (e.g. those published by the Australian Pain Society).
Tolerance, dependence and withdrawal.
Neuroadaptation of the opioid receptors to repeated administration of opioids can produce tolerance and physical dependence. Tolerance is the need for increasing doses to maintain analgesia. Tolerance may occur to both the desired and undesired effects of the opioid.
Physical dependence, which can occur after several days to weeks of continued opioid usage, results in withdrawal symptoms if the opioid is ceased abruptly or the dose is significantly reduced. Withdrawal symptoms can also occur following the administration of an opioid antagonist (e.g. naloxone) or partial agonist (e.g. buprenorphine). Withdrawal can result in some or all of the following symptoms: dysphoria, restlessness/agitation, lacrimation, rhinorrhoea, yawning, sweating, chills, myalgia, mydriasis, irritability, anxiety, increasing pain, backache, joint pain, weakness, abdominal cramps, insomnia, nausea, anorexia, vomiting, diarrhoea, increased blood pressure, increased respiratory rate and increased heart rate. ARX-Oxycodone/Naloxone modified release tablets are not suitable for the treatment of symptoms of opioid withdrawal.
When discontinuing ARX-Oxycodone/Naloxone modified release tablets in a person who may be physically-dependent, the drug should not be ceased abruptly but withdrawn by tapering the dose gradually (see Section 4.4 Special Warnings and Precautions for Use, Ceasing opioids).
In patients undergoing opioid treatment, the switch to ARX-Oxycodone/Naloxone modified release tablets can initially provoke withdrawal symptoms or diarrhoea. These patients require specific attention.
Accidental ingestion/exposure.
Accidental ingestion or exposure of ARX-Oxycodone/Naloxone modified release tablets, especially by children, can result in a fatal overdose of oxycodone. Patients and their caregivers should be given information on safe storage and disposal of unused ARX-Oxycodone/Naloxone modified release tablets (see Section 6.4 Special Precautions for Storage; Section 6.6 Special Precautions for Disposal).
Hyperalgesia.
Hyperalgesia may occur with the use of opioids, particularly at high doses. Hyperalgesia may manifest as an unexplained increase in pain, increased levels of pain with increasing opioid dosages or diffuse sensitivity not associated with the original pain. Hyperalgesia should not be confused with tolerance (see Section 4.4 Special Warnings and Precautions for Use, Tolerance, dependence and withdrawal). If opioid induced hyperalgesia is suspected, the dose should be reduced and tapered off if possible. A change to a different opioid may be required.
Ceasing opioids.
Abrupt discontinuation or rapid decreasing of the dose in a person physically dependent on an opioid may result in serious withdrawal symptoms and uncontrolled pain (see Section 4.4 Special Warnings and Precautions for Use, Tolerance, dependence and withdrawal). Such symptoms may lead the patient to seek other sources of licit or illicit opioids. Opioids should not be ceased abruptly in a patient who is physically dependent but withdrawn by tapering the dose slowly. Factors to take into account when deciding how to discontinue or decrease therapy include the dose and duration of the opioid the patient has been taking, the type of pain being treated and the physical and psychological attributes of the patient. A multimodal approach to pain management should be in place before initiating an opioid analgesic taper. During tapering, patients require regular review and support to manage any increase in pain, psychological distress and withdrawal symptoms.
There are no standard tapering schedules suitable for all patients and an individualised plan is necessary. In general, tapering should involve a dose reduction of no more than 10 percent to 25 percent every 2 to 4 weeks. If the patient is experiencing increased pain or serious withdrawal symptoms, it may be necessary to go back to the previous dose until stable before proceeding with a more gradual taper.
When ceasing opioids in a patient who has a suspected opioid use disorder, the need for medication assisted treatment and/or referral to a specialist should be considered.
Adrenal insufficiency.
Adrenal insufficiency has been reported with opioid use, more often following long-term use. Symptoms may include nausea, vomiting, anorexia, fatigue, weakness, dizziness, or low blood pressure. If adrenal insufficiency is suspected, appropriate laboratory testing is recommended and discontinuation of treatment with ARX-Oxycodone/Naloxone tablets should be considered.
Endocrine effects.
Opioids may influence the hypothalamic-pituitary-adrenal or gonadal axes. Among the changes observed are an increase in serum prolactin and a decrease in levels of cortisol and testosterone. Clinical symptoms may accompany these hormonal changes.
Androgen deficiency may manifest as low libido, impotence, erectile dysfunction, amenorrhea or infertility.
Neonatal withdrawal syndrome.
Chronic use of oxycodone by the mother at the end of pregnancy may result in a withdrawal syndrome (e.g. hypertonia, neonatal tremor, neonatal agitation, myoclonus, convulsions, apnoea or bradycardia) in the neonate. In many reported cases the withdrawal was serious and required treatment. The syndrome is generally delayed for several hours to several days after birth (see Section 4.6 Fertility, Pregnancy and Lactation, Use in pregnancy).
Gastrointestinal toxicity.
Reports of significant oesophageal dysfunction have been observed via high-resolution manometry in patients taking opioid medicines on a long-term basis. Discontinuation or weaning of opioids should be considered in patients presenting with oesophageal complaints including but not limited to dysphagia, regurgitation, or non-cardiac chest pain.
Restless legs syndrome.
Sleep apnoea is more common in patients with restless legs syndrome and caution is advised in treating such patients with ARX-Oxycodone/Naloxone tablets due to the additive risk of respiratory depression.
There is no clinical experience with ARX-Oxycodone/Naloxone tablets in the long-term treatment of RLS beyond 1 year (see Section 4.2 Dose and Method of Administration).
There is no clinical experience of concomitant dopaminergic agents with ARX-Oxycodone/Naloxone modified release tablets in the management of RLS. Additive or synergistic adverse CNS effects such as nausea, dizziness and confusion may occur and the combination was not tested in the clinical trial OXN3502 in RLS patients.
The combination of ARX-Oxycodone/Naloxone tablets with dopaminergic agents for the management of RLS is not recommended.
Clinical abuse potential studies.
1. Study in opioid-dependent subjects.
The likeability of ARX-Oxycodone/Naloxone modified release tablets chewed or intact was compared with oxycodone solution and placebo in a randomised, double-blind, placebo and positive-controlled study in 29 opioid-dependent, methadone-maintained subjects. ARX-Oxycodone/Naloxone, either chewed or intact, was associated with statistically significant lower maximum "Drug Liking" scores (p < 0.001) and statistically significant lower scores for "Take Drug Again" (p < 0.001), compared to oxycodone solution, and was associated with similar mean and median maximum scores for "Drug Liking" and "Take Drug Again", compared to placebo treatment. This indicates that ARX-Oxycodone/Naloxone modified release tablets are expected to result in less potential for abuse by all routes of administration in opioid-dependent subjects compared with immediate release oxycodone.
2. Studies in non-dependent opioid abusers.
Additional studies via the intranasal (IN) and intravenous (IV) routes indicate that ARX-Oxycodone/Naloxone modified release is expected to reduce abuse via the IN and IV routes of administration. ARX-Oxycodone/Naloxone modified release, administered via the IN and IV routes was statistically significantly less preferred over oxycodone HCl powder. No reduction in abuse potential was noted following chewed oral administration in this patient group.
Despite the abuse deterrent properties demonstrated in these studies, abuse and diversion by these and other routes are still possible. As with other opioids, patients should be carefully monitored for signs of abuse and addiction. Abuse or misuse of ARX-Oxycodone/Naloxone modified release tablets by crushing, chewing, snorting, or injecting the dissolved product will result in the uncontrolled delivery of oxycodone and can result in overdose and death.
Formulation.
ARX-Oxycodone/Naloxone modified release tablets must be swallowed whole with sufficient water and must not be broken, chewed or crushed, as this can lead to the rapid release of the active ingredients and absorption of a potentially fatal dose of oxycodone.
ARX-Oxycodone/Naloxone modified release tablets consist of a dual-polymer matrix, intended for oral use only. ARX-Oxycodone/Naloxone modified release tablets contain lactose. Patients with rare hereditary problems of galactose intolerance, Lapp lactase deficiency or glucose-galactose malabsorption should not take ARX-Oxycodone/Naloxone modified release tablets. The empty tablet matrix may be visible in the stool. ARX-Oxycodone/Naloxone modified release tablets may produce positive results in sports agency drug testing procedures.
Use in hepatic impairment.
ARX-Oxycodone/Naloxone modified release tablets should be used with caution in patients with mild hepatic impairment (see Section 5.2 Pharmacokinetic Properties). Whilst the administration of ARX-Oxycodone/Naloxone modified release tablets to these patients does not result in significant levels of oxycodone active metabolites, the plasma concentrations in this patient population may be increased compared with patients having normal hepatic function. Therefore, initiation of dosing in patients with mild hepatic impairment should be reduced to 1/3 to ½ of the usual dose with cautious titration and careful medical monitoring.
Because of the observed increase in naloxone plasma concentrations, and until the clinical relevance of this is established, ARX-Oxycodone/Naloxone modified release tablets are contraindicated in patients with moderate to severe hepatic impairment.
Use in renal impairment.
ARX-Oxycodone/Naloxone modified release tablets should be used with caution in patients with renal impairment (CKD stages 2 to 5) (see Section 5.2 Pharmacokinetic Properties). Whilst the administration of ARX-Oxycodone/Naloxone modified release tablets to these patients does not result in significant levels of oxycodone active metabolites, the plasma concentrations in this patient population may be increased compared with patients having normal renal function. Therefore, initiation of dosing in patients with renal impairment (CKD stages 2 to 5) should be reduced to 1/3 to ½ of the usual dose with cautious titration and careful medical monitoring.
As patients with severe renal impairment (CKD stages 4 and 5) may be at greater risk for opioid withdrawal-related adverse events, consideration should be given to alternative products without naloxone.
Use in the elderly.
As with other opioid initiation and titration, doses in elderly patients who are infirm or debilitated should be reduced to 1/3 to ½ of the usual doses.
The plasma concentrations of oxycodone are only nominally affected by age, being approximately 18% greater in elderly as compared with young subjects. There were no differences in adverse event reporting between young and elderly subjects. The dosage should be adjusted to the intensity of the pain and the sensitivity of the individual patient.
Life-threatening respiratory depression is more likely to occur in elderly, cachectic, or debilitated patients as they may have altered pharmacokinetics or altered clearance compared to younger, healthier patients. Monitor such patients closely, particularly when initiating and titrating ARX-Oxycodone/Naloxone modified release tablets and when ARX-Oxycodone/Naloxone modified release tablets are given concomitantly with other drugs that depress respiration.
As with all opioids, a reduction in dosage may be advisable in hypothyroidism. Exercise caution when administering ARX-Oxycodone/Naloxone modified release tablets to elderly, infirm or debilitated patients, patients with mild hepatic impairment, patients with renal impairment, patients with severely impaired pulmonary function and opioid-dependent patients. Precaution is required in hypotension, hypertension, hypovolaemia, diseases of the biliary tract (e.g. cholelithiasis), pancreatitis, inflammatory bowel disorders, prostatic hypertrophy, adrenocortical insufficiency (Addison's disease), toxic psychosis, myxoedema, opioid-induced paralytic ileus, pre-existing cardiovascular disease and in epileptic disorders or predisposition to convulsions.
As with all opioid preparations, patients who are to undergo cordotomy or other pain-relieving surgical procedures should not receive ARX-Oxycodone/Naloxone modified release tablets for 24 hours before surgery. Pain in the immediate pre-operative period, and any symptoms of opioid withdrawal, should be managed with short-acting analgesic agents. If further treatment with ARX-Oxycodone/Naloxone modified release tablets is then indicated, the dosage should be adjusted to the new post-operative requirement.
ARX-Oxycodone/Naloxone modified release tablets are not recommended for immediate pre-operative use and post-operative use for the first 24 hours after surgery. Depending on the type and extent of surgery, the anaesthetic procedure selected, other co-medication and the individual health status of the patient, the exact timing for initiating treatment with ARX-Oxycodone/Naloxone modified release tablets depends on a careful risk-benefit assessment for each individual patient.
There is no clinical experience in patients with cancer associated with peritoneal carcinomatosis or with sub-occlusive syndrome in advanced stages of digestive and pelvic cancers. Therefore, the use of ARX-Oxycodone/Naloxone modified release tablets in this population is not recommended.
Biliary tract disorders.
Oxycodone can cause an increase in intrabiliary pressure and spasm as a result of its effects on the sphincter of Oddi; therefore, monitor patients with diseases of the biliary tract for worsening symptoms while administering oxycodone. Therefore, ARX-Oxycodone/Naloxone tablets have to be administered with caution in patients with pancreatitis and diseases of the biliary tract.
Paediatric use.
ARX-Oxycodone/Naloxone modified release tablets are not recommended for use in patients under 18 years of age.
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
Alcohol.
Dissolution studies with ARX-Oxycodone/Naloxone modified release tablets were conducted in Standard Gastric Fluid sine pepsin (SGFsp) dissolution media, modified with ethanol at concentrations up to 40% v/v, representative of the most extreme conditions likely to be encountered in vivo. The prolonged release characteristics of ARX-Oxycodone/Naloxone modified release tablets were maintained under these test conditions, and no breakdown of the controlled release mechanism of the formulation was observed.
Anticholinergic agents.
Concurrent use of oxycodone with anticholinergics or medications with anticholinergic activity (e.g. tricyclic antidepressants, antihistamines, antipsychotics, muscle relaxants, anti-Parkinson drugs) may result in increased anticholinergic effects, e.g. an increased risk of severe constipation and/or urinary retention. The presence of naloxone in ARX-Oxycodone/Naloxone modified release tablets, however, may serve to reverse the additive constipative effect, at least in part.
Antihypertensive agents.
Hypotensive effects of these medications may be potentiated when used concurrently with oxycodone, leading to increased risk of orthostatic hypotension.
CNS depressants.
Concurrent use of CNS depressants (including antidepressants, sedatives (incl. benzodiazepines), antipsychotics, hypnotics, general anaesthetics, phenothiazines, other tranquillisers, alcohol, other opioids, gabapentinoids such as pregabalin, anxiolytics, centrally-active anti-emetics, cannabis, anti-histamines, anti-emetics and neuroleptic drugs, etc.) with oxycodone may enhance the CNS-depressant effect resulting in increased risk of respiratory depression, hypotension, profound sedation, coma or death. (see Section 4.4 Special Warnings and Precautions for Use, Risks from concomitant use of benzodiazepines or other CNS depressants, including alcohol).
Intake of alcoholic beverages while being treated with oxycodone should be avoided because this may lead to more frequent undesirable effects such as somnolence and respiratory depression. Oxycodone hydrochloride containing products should be avoided in patients with a history of or present alcohol, drug or medicines abuse.
Serotonin agents.
Concurrent use of oxycodone with serotonin agents, such as a selective serotonin re-uptake inhibitor (SSRI) or a serotonin norepinephrine re-uptake inhibitor (SNRI) may cause serotonin toxicity. The symptoms of serotonin toxicity may include mental-status changes (e.g. agitation, hallucinations, coma), autonomic instability (e.g. tachycardia, labile blood pressure, hyperthermia), neuromuscular abnormalities (e.g. hyperreflexia, incoordination, rigidity), and/or gastrointestinal symptoms (e.g. nausea, vomiting, diarrhoea). Oxycodone should be used with caution and the dosage may need to be reduced in patients using these medications.
Coumarin derivatives.
Opiate agonists have been reported to potentiate the anticoagulant activity of coumarin derivatives. Clinically relevant changes in international normalised ratio (INR or Quick-value) in both directions were observed when oxycodone and coumarin anticoagulants were co-administered.
CYP2D6 and CYP3A4 inhibitors and inducers.
Oxycodone is metabolised in part via the CYP2D6 and CYP3A4 pathways. The activities of these metabolic pathways may be inhibited or induced by various co-administered drugs or dietary elements. ARX-Oxycodone/Naloxone doses may need to be adjusted accordingly.
Drugs that inhibit CYP2D6 activity, such as paroxetine and quinidine, may cause decreased clearance of oxycodone which could lead to an increase in oxycodone plasma concentrations. Concurrent administration of quinidine does not alter the pharmacodynamic effects of oxycodone.
CYP3A4 inhibitors, such as macrolide antibiotics (e.g. clarithromycin), azole-antifungal agents (e.g. ketoconazole), protease inhibitors (e.g. ritonavir), and grapefruit juice may cause decreased clearance of oxycodone which could lead to an increase in oxycodone plasma concentrations.
CYP3A4 inducers, such as rifampin, carbamazepine, phenytoin and St. John's wort, may induce the metabolism of oxycodone and cause increased clearance of the drug, resulting in a decrease in oxycodone plasma concentrations.
Oxycodone metabolism may be blocked by a variety of drugs (e.g. cimetidine, certain cardiovascular drugs and antidepressants), although such blockade has not yet been shown to be of clinical significance with ARX-Oxycodone/Naloxone modified release tablets.
In vitro metabolic studies indicate that no clinically relevant interactions are to be expected between oxycodone and naloxone. At therapeutic concentrations, ARX-Oxycodone/Naloxone modified release tablets are not expected to cause clinically relevant interactions with other concomitantly administered drugs metabolised over the CYP isomers, CYP1A2, CYP2A6, CYP2C9/19, CYP2D6, CYP2E1 and CYP3A4. In addition, the likelihood of clinically relevant interactions between paracetamol, acetylsalicylic acid or naltrexone and the combination of oxycodone and naloxone in therapeutic concentrations is minimal.
In vitro data also suggest that the dopamine agonists, ropinirole, (S)-pramipexole and levodopa have little or no effect on either oxycodone or naloxone major metabolic pathways. Rotigotine had little effect on oxycodone metabolism, and inhibited naloxone metabolism only at concentrations considerably greater than anticipated clinical plasma rotigotine concentrations associated with RLS treatment.
CNS adverse effects associated with dopamine agonists and oxycodone are similar and concurrent use for the treatment of RLS was not assessed in the pivotal RLS study.
Metoclopramide.
Concurrent use with oxycodone may antagonise the effects of metoclopramide on gastrointestinal motility.
Monoamine oxidase inhibitors (MAOIs).
Non-selective MAOIs intensify the effects of opioid drugs which can cause anxiety, confusion and significant respiratory depression. Severe and sometimes fatal reactions have occurred in patients concurrently administered MAOIs and pethidine. Oxycodone should not be given to patients taking non-selective MAOIs or within 14 days of stopping such treatment. As it is unknown whether there is an interaction between selective MAOIs (e.g. selegiline) and oxycodone, caution is advised with this drug combination.
Neuromuscular blocking agents.
Oxycodone may enhance the effects of neuromuscular blocking agents resulting in increased respiratory depression.
Opioid agonist analgesics (including morphine, pethidine).
Additive CNS-depressant, respiratory depressant and hypotensive effects may occur if two or more opioid agonist analgesics are used concurrently.
Opioid agonist-antagonist analgesics (including pentazocine, butorphanol, buprenorphine).
Mixed agonist/antagonist analgesics may reduce the analgesic effect of oxycodone and/or may precipitate withdrawal symptoms.
Dopaminergic agents.
Concomitant use of dopaminergic agents and ARX-Oxycodone/Naloxone modified release tablets for the treatment of RLS is not recommended because efficacy and safety have not been assessed in the clinical study and both medicines are CNS depressants. If dopaminergic agents are used for the management of Parkinson's disease in patients taking ARX-Oxycodone/Naloxone modified release tablets, then the dose of each medicine may need to be reduced.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
No studies have been conducted on the reproductive toxicity of the combination of oxycodone and naloxone. In reproductive toxicology studies of oxycodone alone, no evidence of impaired fertility was seen in male or female rats at oral oxycodone doses of 8 mg/kg/day, approximately half the oxycodone dose at the maximal recommended clinical dose of ARX-Oxycodone/Naloxone modified release tablets, on a body surface area basis. There were also no effects on fertility in rats following oral administration of naloxone at doses up to 800 mg/kg/day, which is about 90-fold the naloxone dose at the maximal recommended clinical dose of ARX-Oxycodone/Naloxone modified release tablets, on a body surface area basis.
No human data on the effect of oxycodone and naloxone on fertility are available.
Despite these fertility studies in animals, prolonged use of opioids may result in impairment of reproductive function, including fertility and sexual dysfunction in both sexes, and irregular menses in women.
(Category C)
ARX-Oxycodone/Naloxone modified release tablets are contraindicated in pregnancy. Oxycodone and naloxone pass into the placenta. There are no adequate and well-controlled studies on the use of ARX-Oxycodone/Naloxone modified release tablets in pregnant women and during childbirth. Long-term administration of oxycodone during pregnancy may lead to withdrawal symptoms in the newborn child, and may cause respiratory depression during childbirth. Infants born to mothers who have received opioids during pregnancy should be monitored for respiratory depression.
No studies have been conducted on the reproductive toxicity of the combination of oxycodone and naloxone. There was no evidence of teratogenicity following oral administration of oxycodone during the period of organogenesis to rats at doses up to 7.2 mg/kg/day (approximately half the oxycodone dose at the maximal recommended clinical dose of ARX-Oxycodone/Naloxone modified release tablets, on a body surface area basis) or to rabbits at doses of up to 112 mg/kg/day (approximately 12-fold the oxycodone dose at the maximal recommended clinical dose of ARX-Oxycodone/Naloxone modified release tablets). There was also no evidence of teratogenicity following oral administration of naloxone during the period of organogenesis to rats and rabbits at respective doses up to 800 and 400 mg/kg/day, which are more than 80-fold the naloxone dose at the maximal recommended clinical dose of ARX-Oxycodone/Naloxone modified release tablets on a body surface area basis.
Because animal reproduction studies are not always predictive of human responses, this medicine should not be used during pregnancy.
ARX-Oxycodone/Naloxone modified release tablets are contraindicated during lactation. Oxycodone passes into breast milk. A milk: plasma ratio of 3.4:1 was measured, and withdrawal symptoms can occur in breastfeeding infants when maternal administration of an opioid analgesic is stopped.
Oral administration of oxycodone to rats from early gestation to weaning did not affect postnatal development parameters at doses up to 6 mg/kg/day (about one-third the oxycodone dose at the maximal recommended clinical dose of ARX-Oxycodone/Naloxone modified release tablets, on a body surface area basis). Oral administration of naloxone to rats from prior to mating to weaning, or from late gestation to weaning, did not affect reproductive or developmental indices up to 800 mg/kg/day (about 90-fold the naloxone dose at the maximal recommended clinical dose of ARX-Oxycodone/Naloxone modified release tablets, on a body surface area basis).
It is not known if naloxone also passes into breast milk. ARX-Oxycodone/Naloxone modified release tablets should not be taken by breastfeeding mothers prior to the infant being weaned.4.7 Effects on Ability to Drive and Use Machines
ARX-Oxycodone/Naloxone modified release tablets may impair the ability to drive and operate machinery, particularly at the commencement of treatment, after dosage increase or opioid rotation, and if ARX-Oxycodone/Naloxone modified release tablets are combined with alcohol or other CNS depressants. The degree of driving impairment can depend upon the dosage and individual susceptibility, and some patients stabilised on a specific dosage may not be affected. All patients should consult with their physician and should not drive or operate machinery if their ability is impaired.
Patients who have experienced somnolence and/or an episode of sudden sleep onset must not drive or operate machinery. Additionally, a dose reduction or termination of therapy may be considered. Because of possible addictive effects, caution should be advised when patients are taking other sedating medicinal products in combination with ARX-Oxycodone/Naloxone (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
4.8 Adverse Effects (Undesirable Effects)
Analgesia.
Adverse drug reactions are typical of full opioid agonists, and tend to reduce with time. The naloxone in ARX-Oxycodone/Naloxone modified release tablets reduces bowel function disorders such as constipation that typically arise during oxycodone analgesic treatment. Anticipation of adverse drug reactions and appropriate patient management can improve acceptability. A reduction in pre-existing laxatives may be appropriate when initiating ARX-Oxycodone/Naloxone modified release tablets in opioid-treated patients.
The following adverse events were reported in the pivotal trials, during the double-blind phase, without attributing causality. See Tables 2, 3 and 4.
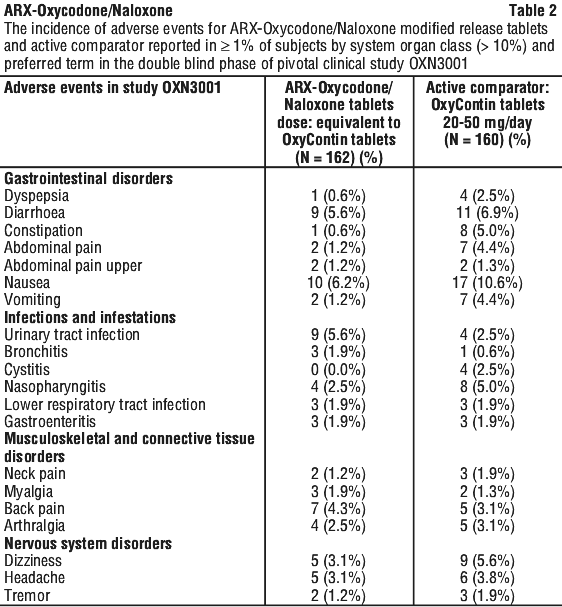
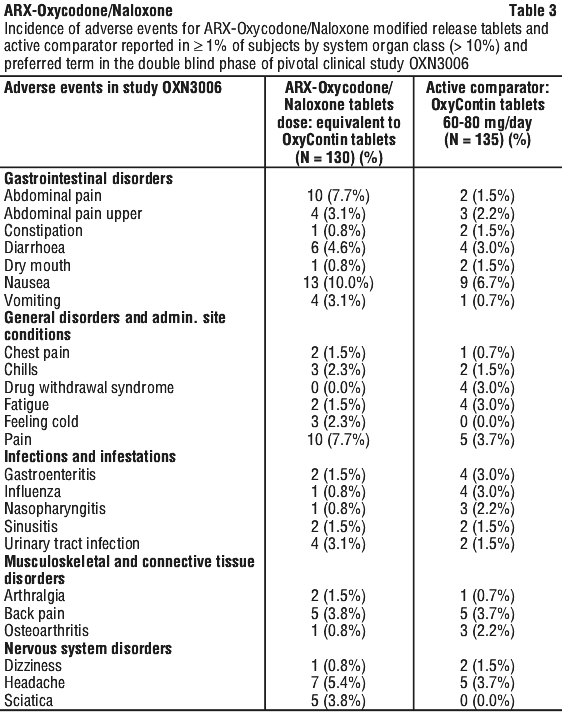
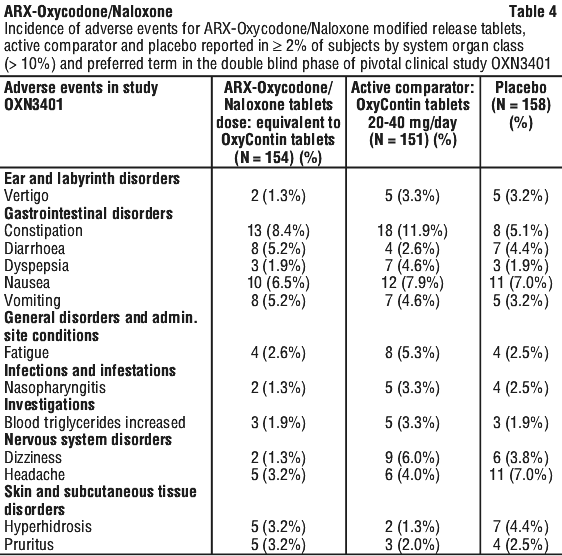 Adverse drug reactions attributable to ARX-Oxycodone/Naloxone modified release tablets were reported at the frequencies below:
Adverse drug reactions attributable to ARX-Oxycodone/Naloxone modified release tablets were reported at the frequencies below:
Very common: ≥ 10%; common: > 1% and < 10%; uncommon: > 0.1% and < 1%; rare: > 0.01% and < 0.1%; very rare: < 0.01%; not known: (cannot be estimated from available data).
The adverse drug reactions listed below are taken cumulatively from clinical trial data and post-marketing data.
Cardiac disorders.
Uncommon: palpitations (in the context of withdrawal symptoms).
Ear and labyrinth disorders.
Common: vertigo.
Endocrine disorders.
Not known: adrenal insufficiency, androgen deficiency.
Eye disorders.
Uncommon: visual impairment.
Gastrointestinal disorders.
Common: abdominal pain, constipation, diarrhoea, dry mouth, dyspepsia, nausea, vomiting.
Uncommon: flatulence.
Not known: eructation, pancreatitis.
General disorders and application site conditions.
Common: asthenic conditions, fatigue.
Uncommon: chest pain, chills, malaise, peripheral oedema, thirst.
Not known: drug withdrawal syndrome.
Hepatobiliary disorders.
Uncommon: hepatic enzymes increased.
Immune system disorders.
Uncommon: hypersensitivity.
Injury, poisoning and procedural complications.
Uncommon: injuries from accidents.
Metabolism and nutrition disorders.
Common: decreased appetite.
Musculoskeletal and connective tissue disorders.
Uncommon: muscle spasms, muscle twitching, myalgia.
Nervous system disorders.
Common: dizziness, headache, somnolence.
Uncommon: disturbance in attention, dysgeusia, speech disorder, tremor, convulsion (particularly in persons with epileptic disorder or predisposition to convulsions), syncope, lethargy.
Not known: sedation, paraesthesia.
Psychiatric disorders.
Common: insomnia.
Uncommon: anxiety, confusional state, depression, nervousness, restlessness, abnormal thinking, libido decreased.
Not known: nightmares, euphoric mood, hallucinations, aggression, drug dependence (see Section 4.4 Special Warnings and Precautions for Use).
Renal and urinary disorders.
Uncommon: micturition urgency.
Not known: urinary retention.
Reproduction system and breast disorders.
Not known: erectile dysfunction.
Respiratory, thoracic and mediastinal disorders.
Uncommon: dyspnoea.
Not known: respiratory depression, central sleep apnoea syndrome.
Skin and subcutaneous tissue disorders.
Common: hyperhidrosis, pruritus, rash.
Vascular disorders.
Common: hot flush.
Uncommon: increase in blood pressure, decrease in blood pressure.
The following additional adverse events are known for oxycodone.
Due to its pharmacological properties, oxycodone may cause respiratory depression, miosis, bronchial spasm, and spasms of non-striated muscles as well as suppress the cough reflex.
Ear and labyrinth disorders.
Uncommon: tinnitus.
Eye disorders.
Uncommon: miosis.
Gastrointestinal disorders.
Common: gastritis, hiccup.
Uncommon: colic, dysphagia, gastrointestinal disorder, ileus, stomatitis.
Not known: dental caries.
General disorders and administration site conditions.
Common: fever.
Uncommon: facial flushing, lymphadenopathy, neck pain, oedema.
Not known: drug withdrawal syndrome neonatal, drug tolerance.
Hepatobiliary disorders.
Uncommon: biliary spasm.
Not known: cholestasis, sphincter of Oddi dysfunction.
Immune system disorders.
Uncommon: allergic reaction, anaphylactoid reaction.
Not known: anaphylactic reaction.
Metabolism and nutrition disorders.
Uncommon: dehydration, hyponatraemia.
Rare: increased appetite.
Musculoskeletal and connective tissue disorders.
Uncommon: involuntary muscle contractions, muscular rigidity.
Nervous system disorders.
Common: faintness.
Uncommon: amnesia, drowsiness, gait abnormal, hyperkinesia, hypertonia, hypoaesthesia, hypothermia, raised intracranial pressure, stupor.
Not known: hyperalgesia.
Psychiatric disorders.
Common: mood changes.
Uncommon: agitation, affect lability, disorientation, dysphoria.
Renal and urinary disorders.
Common: ureteric spasm, urinary abnormalities, urinary tract infection.
Reproductive system and breast disorders.
Uncommon: hypogonadism.
Not known: amenorrhoea.
Respiratory, thoracic and mediastinal disorders.
Common: bronchospasm, pharyngitis, voice alteration.
Skin and subcutaneous tissue disorders.
Uncommon: dry skin, exfoliative dermatitis.
Rare: urticaria.
Vascular disorders.
Common: orthostatic hypotension.
Uncommon: migraine, vasodilatation.
Drug dependence.
The frequencies above regarding drug dependence, drug withdrawal syndrome and drug tolerance reflects that although risk is low with short term and low dose use, it is highly variable.
Repeated use of ARX-Oxycodone/Naloxone can lead to drug dependence, even at therapeutic doses. The risk of drug dependence may vary depending on a patient's individual risk factors, dosage, and duration of opioid treatment (see Section 4.4 Special Warnings and Precautions for Use).
Management of common adverse effects.
If nausea and vomiting are troublesome, oxycodone may be combined with an antiemetic. Constipation must be treated with appropriate laxatives. Overdose may produce respiratory depression. Compared with other opioids, oxycodone is associated with low histamine release although urticaria and pruritus may occur.
Restless legs syndrome.
Adverse drug reactions reported in clinical Study OXN3502 are consistent with the expected safety profile of opioid analgesics. These adverse events are not unexpected in a study of an active opioid treatment and inactive placebo, and consistent with observations from studies of dopaminergic agents versus placebo in RLS.
Adverse drug reactions associated with ARX-Oxycodone/Naloxone modified release tablets in pain and not observed in RLS study population were added with the frequency of not known. See Table 5.
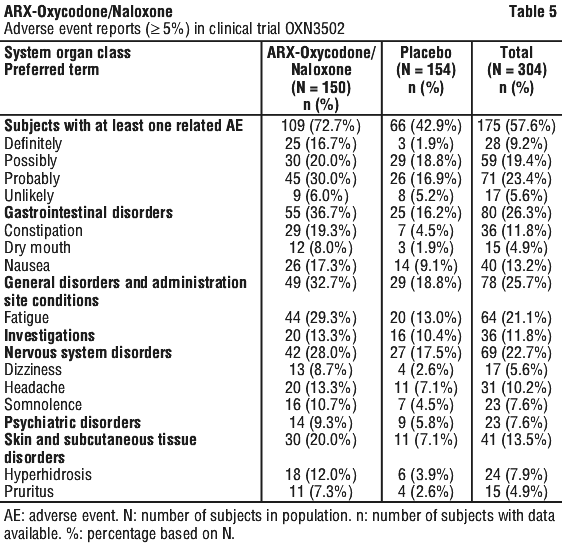
Immune system disorders.
Not known: hypersensitivity.
Metabolism and nutrition disorders.
Common: decreased appetite up to loss of appetite.
Psychiatric disorders.
Common: insomnia, depression.
Uncommon: libido decreased, sleep attacks.
Not known: abnormal thinking, anxiety, confusion, nervousness, restlessness, euphoric mood, hallucination, nightmares.
Nervous system disorders.
Very common: Headache, somnolence.
Common: dizziness, disturbance in attention, tremor, paraesthesia.
Uncommon: dysgeusia.
Not known: convulsions (particularly in persons with epileptic disorder or predisposition to convulsions), sedation, speech disorder, syncope.
Eye disorders.
Common: visual impairment.
Ear and labyrinth disorders.
Common: vertigo.
Cardiac disorders.
Not known: angina pectoris in particular in patients with history of coronary artery disease, palpitations, tachycardia.
Vascular disorders.
Common: hot flush, blood pressure decreased, blood pressure increased.
Respiratory, thoracic and mediastinal disorders.
Uncommon: dyspnoea.
Not known: cough, rhinorrhoea, respiratory depression, yawning.
Gastrointestinal disorders.
Very common: constipation, nausea.
Common: abdominal pain, dry mouth, vomiting.
Uncommon: flatulence.
Not known: abdominal distension, diarrhoea, dyspepsia, eructation, tooth disorder.
Hepatobiliary disorders.
Common: hepatic enzymes increased (alanine aminotransferase increased, gamma-glutamyltransferase increased).
Not known: biliary colic.
Skin and subcutaneous tissue disorders.
Very common: hyperhidrosis.
Common: pruritus, skin reactions.
Musculoskeletal and connective tissue disorders.
Not known: muscle spasms, muscle twitching, myalgia.
Renal and urinary disorders.
Not known: micturition urgency, urinary retention.
Reproductive system and breast disorders.
Uncommon: erectile dysfunction.
General disorders and administration site conditions.
Very common: fatigue.
Common: chest pain, chills, thirst, pain.
Uncommon: drug withdrawal syndrome, oedema peripheral.
Not known: malaise.
Investigation.
Not known: weight decreased, weight increased.
Injury, poisoning and procedural complications.
Uncommon: injuries from accidents.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/safety/reporting-problems http://www.tga.gov.au/reporting-problems.4.9 Overdose
Depending upon the history of the patient, an overdose of ARX-Oxycodone/Naloxone modified release tablets may be manifested by symptoms triggered by oxycodone (opioid receptor agonist) or by naloxone (opioid receptor antagonist). However, symptoms of naloxone overdosage are unlikely (treat symptomatically in a closely-supervised environment).
Symptoms of oxycodone overdosage.
Acute overdose with oxycodone can be manifested by miosis (dilated if hypoxia is severe), cold and/or clammy skin, respiratory depression (reduced respiratory rate and/or tidal volume, cyanosis), extreme somnolence progressing to stupor or coma, hypotonia, bradycardia and hypotension. Toxic leukoencephalopathy has been observed with oxycodone overdose. Coma, non-cardiogenic pulmonary oedema and circulatory failure may occur in more serious cases and may lead to a fatal outcome.
The features of overdosage may be delayed with a controlled release product such as ARX-Oxycodone/Naloxone modified release tablets.
Treatment of oxycodone overdosage.
Primary attention should be given to immediate supportive therapy with the establishment of adequate respiratory exchange through the provision of a patent airway and institution of assisted or controlled ventilation. Adequate body temperature and fluid balance should be maintained.
Oxygen, intravenous fluids, vasopressors, infusions and other supportive measures should be employed, as necessary, to manage the circulatory shock accompanying an overdose. Cardiac arrest or arrhythmias may require cardiac massage or defibrillation. Artificial ventilation should be applied if necessary and fluid and electrolyte metabolism maintained.
Activated charcoal may reduce absorption of the drug if given within one to two hours after ingestion. Administration of activated charcoal should be restricted to patients who are fully conscious with an intact gag reflex or protected airway. A saline cathartic or sorbitol added to the first dose of activated charcoal may speed gastrointestinal passage of the product. In patients who are not fully conscious or have an impaired gag reflex, consideration should be given to administering activated charcoal via a nasogastric tube, once the airway is protected.
Whole bowel irrigation (e.g. 1 or 2 litres of polyethylene glycol solution orally per hour until rectal effluent is clear) may be useful for gut decontamination. Whole bowel irrigation is contraindicated in patients with bowel obstruction, perforation, ileus, haemodynamic instability or compromised, unprotected airways and should be used cautiously in debilitated patients and where the condition may be further compromised. Concurrent administration of activated charcoal and whole bowel irrigation may decrease the effectiveness of the charcoal (there may be competition for the charcoal binding site between the polyethylene glycol and the ingested drugs) but the clinical relevance is uncertain. Prolonged periods of observation (days) may be required for patients who have overdosed with long-acting preparations.
If there are signs of clinically significant respiratory or cardiovascular depression, an opioid antagonist should be considered. Naloxone hydrochloride at a dose of 0.4-2 mg intravenously is a specific antidote for respiratory depression due to overdosage or as a result of unusual sensitivity to oxycodone (please refer to naloxone product information for further information). Concomitant efforts at respiratory resuscitation should be carried out. Administration of naloxone should be repeated at 2-3 minute intervals, as clinically necessary. An infusion of 2 mg naloxone in 500 mL of 0.9% sodium chloride or 5% dextrose (0.004 mg/mL naloxone), run at a rate aligned to previously administered bolus doses and to the patient's response, is also a possible alternative.
The duration of action of oxycodone may exceed that of the antagonist. Consequently, the patient should remain under continued surveillance and dosing of the antagonist continued as needed to maintain adequate respiration.
In an individual physically dependent on opioids, administering opioid antagonists may precipitate a withdrawal syndrome and should be avoided if possible. Withdrawal syndrome may lead to agitation, hypertension, tachycardia and risk of vomiting with possible aspiration. The severity of withdrawal depends on the degree of dependence and the antagonist dose. If required for serious respiratory depression, the antagonist should be administered with extreme care, commencing with 10 to 20% of the usual recommended initial dose and titrating.
Toxicity.
Due to the great interindividual variation in sensitivity to opioids it is difficult to determine an exact dose of any opioid that is toxic or lethal. Crushing and taking the contents of a controlled release dosage form leads to the release of oxycodone in an immediate fashion; this might result in a fatal overdose. The toxic effects and signs of overdosage may be less pronounced than expected, when pain and/or tolerance are manifest.
For information on the management of overdose, contact the Poisons Information Centre on 13 11 26 (Australia).5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Mechanism of action.
Oxycodone is a full opioid receptor agonist whose principal therapeutic action is analgesia. It has an affinity for endogenous mu, kappa and delta opiate receptors in the brain, spinal cord and peripheral organs (e.g. intestine). Binding of oxycodone to endogenous opioid receptors in the central nervous system (CNS) results in pain relief. Oxycodone is similar to morphine in its action. Other pharmacological actions of oxycodone are in the CNS (respiratory depression, antitussive, anxiolytic, sedative and miosis), smooth muscle (constipation, reduced gastric, biliary and pancreatic secretions, sphincter of Oddi spasm and transient elevations in serum amylase), and cardiovascular system via histamine release and peripheral vasodilation (pruritus, flushing, red eyes, sweating and orthostatic hypotension).
Non-clinical studies have demonstrated differing immunomodulatory effects of naturally occurring opioids e.g. morphine, codeine. The clinical significance of these findings is not known. It is not known whether oxycodone, a semi-synthetic opioid, has similar effects.
Naloxone also has an affinity for endogenous opiate receptors in the brain, spinal cord and peripheral organs (e.g. intestine). However, in contrast to oxycodone, naloxone is a competitive opioid antagonist at opiate receptors, which can prevent or reverse the effects of opioid agonists.
Naloxone reduces bowel function disorders such as constipation that typically arise during opioid analgesic treatment with e.g. oxycodone, due to its local competitive antagonism of the opioid receptor-mediated oxycodone effect in the gut. Diarrhoea may be a possible effect of naloxone, especially at the beginning of treatment, and tends to be transient. Oral administration of naloxone is unlikely to result in a clinically relevant systemic effect due to a pronounced first-pass effect and its very low oral bioavailability upon oral administration (< 3%).
Clinical trials.
Analgesia. 1. Study 3001.
This 12-week randomised, double-blind, parallel-group study, in patients with non-malignant pain experiencing opioid-induced constipation, assessed constipation symptoms (as measured by the Bowel Function Index [BFI]) in patients taking ARX-Oxycodone/Naloxone modified release tablets compared with those taking oxycodone controlled release (CR) tablets. 272 patients were randomised to the double-blind phase (136 in each group), with the oxycodone dose between 20-50 mg/day. A secondary objective was to estimate the average pain over the last 24 hours (as measured by the pain intensity scale) at each double-blind visit.
Patients in the ARX-Oxycodone/Naloxone modified release tablets group showed an improved bowel function compared with those on oxycodone CR tablets from one week after the start of the double-blind phase (Visit 4), continuing until the end of the study (Visit 8). Statistical significance was seen by four weeks/Visit 6 (15.2; p < 0.0001; CI -18.2 to -12.2). The mean pain intensity scores for average pain over the last 24 hours were comparable between the two groups, which was maintained until the end of the study with no significant treatment differences seen (0.014; 95% CI; -0.2026 to 0.2304). The safety profile of ARX-Oxycodone/Naloxone modified release tablets is consistent with those of other strong opioids.
2. Study 3006.
This 12-week randomised, double-blind, parallel-group study, in patients with non-malignant pain experiencing opioid-induced constipation, also assessed constipation symptoms (measured by BFI) in patients taking ARX-Oxycodone/Naloxone modified release tablets compared with those taking oxycodone CR tablets. 278 patients were randomised to the double-blind phase (130 on ARX-Oxycodone/Naloxone modified release tablets, 135 on oxycodone CR tablets, 13 were excluded because of study questionnaire irregularities), and the oxycodone dose for each group was between 60 and 80 mg/day.
Throughout the first 4 weeks of the double-blind phase (Visits 3-6), the difference between the mean BFI scores for the two groups was statistically significant in favour of ARX-Oxycodone/Naloxone modified release tablets (-14.9; p < 0.0001; CI -17.9 to -11.9). The actual observed difference of the means was -12.3 (ARX-Oxycodone/Naloxone modified release tablets 40.94; oxycodone CR 53.27). Patients in the ARX-Oxycodone/Naloxone modified release tablets group had a reduced mean observed BFI score from one week after randomisation into the double-blind phase (Visit 4), continuing to the end of the study (Visit 8), but this was not seen for the oxycodone CR tablet group. The mean pain intensity scores for average pain over the last 24 hours were comparable between the groups at baseline (Visit 3), and this was maintained throughout the double-blind phase until the end of the study (Visit 8), with no significant treatment differences seen between the two groups (model estimated treatment difference: 0.010; 95% CI; -0.14 to 0.34). The safety profile of ARX-Oxycodone/Naloxone modified release tablets is consistent with those of other strong opioids.
3. Study OXN1006.
This open-label, single-dose, parallel-group study, compared the pharmacokinetics of oxycodone and naloxone from an oxycodone/naloxone (OXN) prolonged-release (PR) tablet 10/5 mg in patients with varying degrees of hepatic impairment and healthy volunteers.
Significant differences in pharmacokinetic parameters between subjects with hepatic impairment (rated as mild, moderate or severe) and healthy volunteers were seen as summarised in Table 6 (values indicate % of healthy volunteer result):
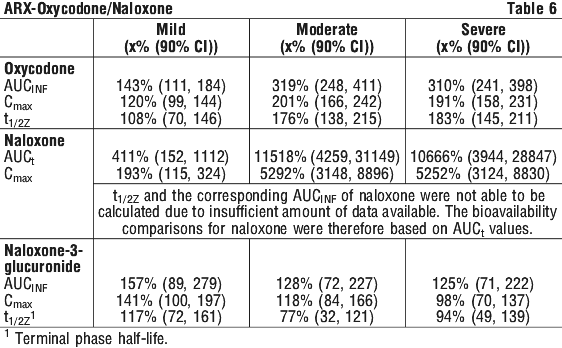
4. Study OXN1007.
This open-label, single-dose, parallel-group study, compared the pharmacokinetics of oxycodone and naloxone from an oxycodone/naloxone (OXN) prolonged release (PR) tablet 10/5 mg in patients with varying degrees of renal impairment and healthy volunteers.
Significant differences in pharmacokinetic parameters between subjects with renal impairment (rated as mild, moderate or severe) and healthy volunteers were seen as summarised in Table 7 (values indicate % of healthy volunteer result):
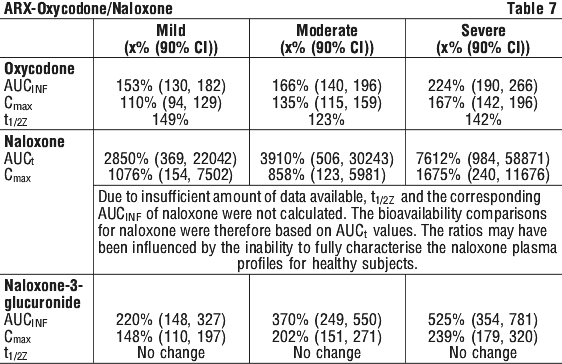
5. Study OXN3506.
The efficacy of ARX-Oxycodone/Naloxone doses up to 160/80 mg daily was assessed in a randomised, double-blind, double-dummy, parallel-group, multiple-dose study in 243 patients with non-malignant or malignant pain requiring high doses of opioids and suffering from constipation caused/ aggravated by opioids. Patients were treated with ARX-Oxycodone/Naloxone tablets (in the range of 50/25 to 80/40 mg twice daily) or oxycodone controlled release (CR) tablets (in the range of 50-80 mg twice daily) for up to 5 weeks. The primary objectives were to demonstrate that subjects taking ARX-Oxycodone/Naloxone tablets have improvement in symptoms of constipation as measured by the bowel function index (BFI) compared to subjects taking oxycodone CR tablets alone, and to demonstrate non-inferiority of ARX-Oxycodone/Naloxone tablets compared to oxycodone CR tablets with respect to the analgesic efficacy based on the subject's 'Average pain over last 24 Hours'. See Table 8.
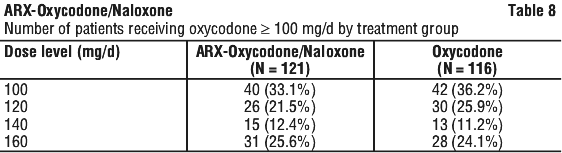 The results show a clinically relevant and statistically significant improvement of the BFI scores in the ARX-Oxycodone/Naloxone group compared to oxycodone CR tablets. The improvements consistently appeared in the full analysis (FA) as well as the per protocol (PP) population, in all the subgroups, in sensitivity analysis with last observation carried forward (LOCF) imputation, and in all 3 single BFI parameters. See Table 9.
The results show a clinically relevant and statistically significant improvement of the BFI scores in the ARX-Oxycodone/Naloxone group compared to oxycodone CR tablets. The improvements consistently appeared in the full analysis (FA) as well as the per protocol (PP) population, in all the subgroups, in sensitivity analysis with last observation carried forward (LOCF) imputation, and in all 3 single BFI parameters. See Table 9.
 In the FA population at week 1 the mean BFI decreased by -28.3 in the ARX-Oxycodone/Naloxone group and by -13.1 in the oxycodone CR tablets group. A median decrease of -23.3 in the ARX-Oxycodone/Naloxone group compared with -6.7 in the oxycodone CR tablets group was observed.
In the FA population at week 1 the mean BFI decreased by -28.3 in the ARX-Oxycodone/Naloxone group and by -13.1 in the oxycodone CR tablets group. A median decrease of -23.3 in the ARX-Oxycodone/Naloxone group compared with -6.7 in the oxycodone CR tablets group was observed.
At week 5, the mean BFI scores for the two groups was statistically significant (p < 0.001, CI: -22.23, -9.86) and clinically relevant in favour of ARX-Oxycodone/Naloxone group (mean difference -16.05 ± 3.14)) and an improvement in BFI was confirmed with ARX-Oxycodone/Naloxone compared with oxycodone CR tablets (p < 0.001, CI: -20.60, -8.40).
The pain value at the beginning of the double-blind phase served as the baseline value. No clinically relevant change to baseline was observed throughout the double-blind phase. At week 5 the mean change to baseline was 0.1 in the ARX-Oxycodone/Naloxone group and 0.0 in the oxycodone CR tablets group.
The average pain intensity over the last 24 hours was comparable between the two groups and was maintained until the end of the study. ARX-Oxycodone/Naloxone was not more than 20% less effective than oxycodone CR alone in providing analgesia (p < 0.001).
Restless legs syndrome. Study OXN3502.
This 12-week randomised, double-blind placebo-controlled, parallel-group, multicentre study, assessed the efficacy and safety in the symptomatic treatment of patients with moderate to severe idiopathic RLS with daytime symptoms and an inadequate response to dopaminergic treatment. Dopaminergic agents were not permitted during the study.
The study comprised a pre-randomisation phase of up to 24 days (including a wash-out period of 7-10 days), a double-blind treatment phase of 12 weeks, and an open-label extension phase of 40 weeks.
The study's primary objective was to demonstrate superior efficacy of ARX-Oxycodone/Naloxone compared to placebo in the improvement of symptom severity of RLS as measured by the International Restless Legs Syndrome Study Group Rating Scale total score (IRLS scale).
IRLS scale: 0 to 10 = mild; 11 to 20 = moderate; 21 to 30 = severe; 31 to 40 = very severe). Patients commencing treatment in this study had severe to very severe disease with median IRLS of 33 (Range 21 to 41).
The primary endpoint was the change in the IRLS score from baseline (Visit 3) to the final maintenance period assessment.
The secondary efficacy endpoints were scores measures of clinical global impression (CGI), RLS-6-rating scale, pain-numeric rating scale (NRS) and the quality of life (QoL).
The 132 patients were initially treated with 5 mg oxycodone hydrochloride/ 2.5 mg naloxone hydrochloride twice daily, but up-titrated to higher dose levels of ARX-Oxycodone/Naloxone tablets (10/5 mg, 20/10 mg and 40/20 mg twice daily) if needed. Significant improvement of RLS during the entire treatment period was shown with a decrease in the mean IRLS score of 8.15 points with a statistically significant difference of 95% CI:5.46, 10.85, p < 0.001; compared to placebo (n = 144) at week 12.
The onset of efficacy was demonstrated from as early as week 1 of treatment, with a decrease in the mean IRLS score of more than 10 points between baseline and week 1. Similar results were shown for the RLS symptom severity improvement (as measured by the RLS-6-rating scale), in quality of life as measured by a QoL-RLS questionnaire, in sleep quality (measured by MOS sleep scale), and for the proportion of IRLS score remitters. No subject had a confirmed case of augmentation during the study.
Primary efficacy results presented by IRLS sum score are summarised in Table 10.
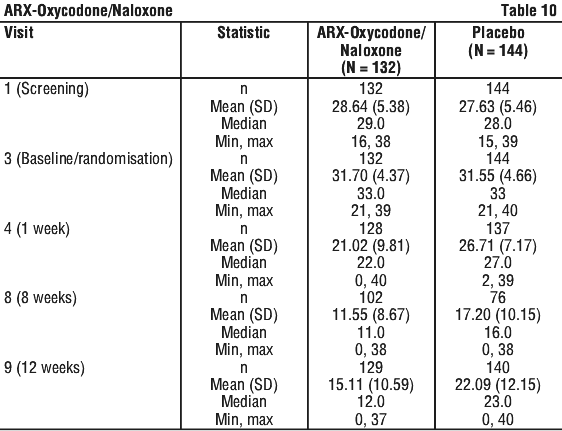
5.2 Pharmacokinetic Properties
The pharmacokinetic characteristics of oxycodone from ARX-Oxycodone/Naloxone modified release tablets are comparable to those from controlled release OxyContin tablets and demonstrate bioequivalence between these two long-acting oxycodone formulations. In addition, dose proportionality has been established for the ARX-Oxycodone/Naloxone 5/2.5 mg, 10/5 mg, 15/7.5 mg, 20/10 mg, 30/15 mg, 40/20 mg, 60/30 mg and 80/40 mg modified release tablet strengths for both peak plasma concentrations (Cmax) and extent of absorption (AUC) facilitating reliable dose titration and interchangeability between tablet strengths.
Absorption.
Oxycodone has a mean bioavailability of approximately 50% following oral administration in healthy volunteers. In cancer patients, an oral bioavailability of up to 87% has been reported.
Following absorption, oxycodone is distributed throughout the body. Approximately 45% is bound to plasma protein.
In a study of ARX-Oxycodone/Naloxone modified release tablets in elderly subjects (≥ 65 years), plasma concentrations of oxycodone were only nominally affected by age, being approximately 18% greater in elderly compared with young subjects.
Female subjects have, on average, plasma oxycodone concentrations up to 25% higher than males on a bodyweight-adjusted basis.
Following ingestion of a high-fat breakfast, the maximum plasma concentration (Cmax) and bioavailability of oxycodone from ARX-Oxycodone/Naloxone modified release tablets were nominally increased compared with fasting state administration but were not considered clinically relevant. ARX-Oxycodone/Naloxone modified release tablets may be taken with or without food.
Following ingestion, oral naloxone is subject to a significant first-pass metabolism and its oral bioavailability is less than 3%.
Metabolism.
Oxycodone has an elimination half-life of approximately 4.8 hours and is metabolised principally in the liver via CYP3A4 and CYP2D6 to noroxycodone, oxymorphone, noroxymorphone, 6α and β oxycodol and conjugated glucuronides. Oxymorphone and noroxymorphone have some analgesic activity. However, oxymorphone is present in plasma at low concentrations and noroxymorphone, due to its low lipophilicity, does not penetrate the blood-brain barrier to a significant extent. Consequently, the contribution of these metabolites to the overall analgesic effect is insignificant. Oxycodone and its metabolites are excreted in urine and faeces.
After parenteral administration, naloxone has a plasma half-life of approximately one hour. Naloxone is metabolised in the liver to its principal metabolites, naloxone glucuronide, 6β-naloxol and its glucuronide, and excreted in the urine.
Impaired hepatic function.
A study has shown that plasma concentrations of both oxycodone and naloxone are elevated in patients with hepatic impairment. Naloxone plasma concentrations were affected to a greater extent than oxycodone. The clinical relevance of a relatively high naloxone exposure in hepatically impaired patients is not yet known. Caution must be exercised in administering ARX-Oxycodone/Naloxone modified release tablets to patients with mild hepatic impairment. ARX-Oxycodone/Naloxone modified release tablets are contraindicated in patients with moderate to severe hepatic impairment.
Impaired renal function.
A study has shown that plasma concentrations of both oxycodone and naloxone are elevated in patients with renal impairment. Naloxone plasma concentrations were affected to a greater extent than oxycodone. The clinical relevance of a relatively high naloxone exposure in renally impaired patients is not yet known. Caution should be exercised when administering ARX-Oxycodone/Naloxone modified release tablets to patients with renal impairment (see Section 4.4 Special Warnings and Precautions for Use).
5.3 Preclinical Safety Data
Genotoxicity.
The results of in vitro and in vivo studies indicate that the genotoxic risk of oxycodone to humans is minimal or absent at the systemic oxycodone concentrations that are achieved therapeutically. Oxycodone showed mutagenic activity in a mouse lymphoma assay, but was inactive in bacterial gene mutation assays. It also induced chromosomal aberrations in human lymphocytes in vitro, but not in immature erythrocytes in vivo in mice. Similar to oxycodone, naloxone induced gene mutations and chromosomal aberrations in mouse lymphoma cell lines and human lymphocytes in vitro, respectively, but did not induce chromosomal aberrations in immature erythrocytes under in vivo conditions.
Carcinogenicity.
Long-term studies in animals to evaluate the carcinogenic potential of oxycodone/naloxone in combination have not been conducted.
Carcinogenicity was evaluated in a 2-year oral gavage study conducted in Sprague-Dawley rats. Oxycodone did not increase the incidence of tumours in male and female rats at doses up to 6 mg/kg/day (equivalent to 6.8 mg/day in men and 24.6 mg/day in women, based on estimated AUC values). The doses were limited by opioid-related pharmacological effects of oxycodone.
Naloxone was not carcinogenic in a 24-month dietary study in rats at doses up to 100 mg/kg/day, which is about 11-fold the naloxone dose at the maximal recommended clinical dose of ARX-Oxycodone/Naloxone modified release tablets, on a body surface area basis.6 Pharmaceutical Particulars
6.1 List of Excipients
See Section 2 Qualitative and Quantitative Composition.
6.2 Incompatibilities
Incompatibilities were not assessed as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Store below 25°C.
6.5 Nature and Contents of Container
ARX-Oxycodone/Naloxone modified release tablets are presented in PVC/Al blister packs. Not all pack sizes are available in Australia:
2.5/1.25 mg.
20, 28 and 60 tablets in blister packs.
5/2.5 mg.
20, 28 and 60 tablets in blister packs.
10/5 mg.
20, 28 and 60 tablets in blister packs.
15/7.5 mg.
20, 28 and 60 tablets in blister packs.
20/10 mg.
20, 28 and 60 tablets in blister packs.
30/15 mg.
20, 28 and 60 tablets in blister packs.
40/20 mg.
20, 28 and 60 tablets in blister packs.
60/30 mg.
20, 28 and 60 tablets in blister packs.
80/40 mg.
20, 28 and 60 tablets in blister packs.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Oxycodone hydrochloride is a white, crystalline, odourless powder readily soluble in water, sparingly soluble in ethanol and nearly insoluble in ether.
Naloxone hydrochloride dihydrate is an off-white powder soluble in water.
Oxycodone hydrochloride.
The chemical name is 4,5a-epoxy-14-hydroxy-3-methoxy-17-methylmorphinan-6-one hydrochloride. The molecular formula is C18H21NO4.HCl and molecular weight is 351.83. The pKa is 8.9 and the Partition Coefficient Log P is 0.7.
Naloxone hydrochloride.
The chemical name is 17-allyl-4,5a-epoxy-3,14-dihydroxymorphinan-6-one hydrochloride dihydrate. It is a synthetic congener of oxymorphone, with molecular formula C19H21NO4.HCl.2.H2O and molecular weight 399.87. The pKa is 7.9 and the Partition Coefficient Log P is 1.5.
Chemical structure.
The structural formula for oxycodone hydrochloride is:
 The structural formula for naloxone hydrochloride dihydrate is:
The structural formula for naloxone hydrochloride dihydrate is:

CAS number.
Oxycodone hydrochloride.
124-90-3.
Naloxone hydrochloride.
51481-60-8.7 Medicine Schedule (Poisons Standard)
S8.
Summary Table of Changes