1 Name of Medicine
Methylphenidate hydrochloride.
2 Qualitative and Quantitative Composition
Attora LA modified release capsules contain 10 mg, 20 mg, 30 mg, 40 mg or 60 mg of methylphenidate hydrochloride.
List of excipients with known effect - Contains sulfites.
For the full list of the excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Attora LA are modified release capsules that presented as follows:
10 mg capsules.
Opaque hard gelatin capsule with rich yellow cap and white body imprinted "10" with black ink filled with white to off-white spherical pellets.
20 mg capsules.
Opaque hard gelatin capsule with white cap and white body imprinted "20" with black ink filled with white to off-white spherical pellets.
30 mg capsules.
Opaque hard gelatin capsule with ivory cap and ivory body imprinted "30" with black ink filled with white to off-white spherical pellets.
40 mg capsules.
Opaque hard gelatin capsule with rich yellow cap and rich yellow body imprinted "40" with black ink filled with white to off-white spherical pellets.
60 mg capsules.
Opaque hard gelatin capsule with ivory cap and white body imprinted "60" with black ink filled with white to off-white spherical pellets.4.1 Therapeutic Indications
Attora LA capsules are indicated for the treatment of ADHD.
Attention-deficit hyperactivity disorder (ADHD).
ADHD was previously known as attention-deficit disorder. Other terms used to describe this behavioural syndrome include: minimal brain dysfunction in children, hyperkinetic child syndrome, minimal brain damage, minimal cerebral dysfunction, minor cerebral dysfunction and psycho-organic syndrome of children.
Attora LA are indicated as an integral part of a total treatment program for ADHD that may include other measures (psychological, educational and social) for patients with this syndrome. Stimulants are not intended for use in the patient who exhibits symptoms secondary to environmental factors and/or other primary psychiatric disorders, including psychosis.
Special diagnostic considerations for ADHD in children.
The aetiology of this syndrome is unknown and there is no single diagnostic test. Adequate diagnosis requires the use, not only of medical, but also of psychological, educational and social resources. Characteristics commonly reported include: chronic history of short attention span, distractibility, emotional lability, impulsivity, moderate to severe hyperactivity, minor neurological signs and an abnormal EEG. Learning may or may not be impaired. The diagnosis must be based upon a complete history and evaluation of the child and not solely on the presence of one or more of these characteristics.
Drug treatment is not indicated for all children with this syndrome. Stimulants are not intended for use in children who exhibit symptoms secondary to environmental factors (e.g. child abuse in particular) or primary psychiatric disorders. Appropriate educational placement is essential and psychosocial intervention is generally necessary. When remedial measures alone are insufficient, the decision to prescribe stimulant medicine will depend upon the physician's assessment of the chronicity and severity of the child's symptoms.
Continuation of treatment in adolescent and special diagnostic considerations for ADHD in adults.
There is limited information to guide clinicians about how long older adolescents should continue to receive treatment with drugs for attention deficit hyperactivity disorder (ADHD). The decision should be based on the extent to which symptoms of ADHD and social functioning have improved to a point that medication is no longer needed. If older adolescents have been largely symptom-free for a year and are functioning well, a trial without medication is warranted. This should be undertaken at times of low stress such as during holidays or in a period when a school routine is well established.
ADHD needs to be considered in adults who present with longstanding symptoms suggestive of ADHD (inattention, impulsivity, disorganisation) that appear to have started in childhood and are persisting into adult life. Further, people with personality disorder and/or problems with drug use accompanied by a significant level of impulsivity and inattention should be referred for evaluation by a psychiatrist with the training and skills required to assess and treat ADHD. This expertise is necessary due to the overlap of ADHD symptoms with anxiety, mood and personality disorders.4.2 Dose and Method of Administration
Pre-treatment screening.
Treatment should only be initiated by specialist physicians with experience in the use of the drug. Before initiating Attora LA treatment, patients should be assessed for pre-existing cardiovascular and psychiatric disorders and a family history of sudden death, ventricular arrhythmia and psychiatric disorders. Weight and height should also be measured before treatment and documented on a growth chart (see Section 4.3 Contraindications; Section 4.4. Special Warnings and Precautions for Use).
Dosage.
The dosage of Attora LA should be individualised according to the patient's clinical needs and responses.
Treatment with Attora LA should be initiated at a low dose, with increments at weekly intervals.
ADHD.
In the treatment of ADHD, an attempt should be made to time administration of the drug to coincide with periods of greatest academic, behavioural or social difficulties for the patient.
If symptoms do not improve after dose titration over a one-month period, the drug should be discontinued.
If symptoms worsen or other adverse effects occur, the dosage should be reduced or, if necessary, the drug discontinued.
If the effect of the drug wears off too early in the evening, disturbed behaviour and/or inability to go to sleep may recur. A small evening dose of methylphenidate 10 mg immediate release tablet may help solve this problem.
* Methylphenidate 10 mg immediate release tablets are unavailable in this brand, however, is available in other brands. Refer to the specific product information for this formulation for its complete dosage and administration instructions.
Periodic assessment of the treatment in ADHD. Drug treatment does not need to be indefinite. Physicians should periodically re-evaluate the treatment with trial periods off medication to assess the patient's functioning without pharmacotherapy. Improvement may be sustained when the drug is either temporarily or permanently discontinued. When used in children with ADHD, treatment can usually be discontinued during or after puberty.
If therapy is interrupted for reasons other than those stated above, it should not be restarted at the dose that had been reached prior to treatment interruption, but should be re-titrated.
Children and adolescents (6 years and over). Maximum daily doses.
Daily doses above 60 mg are not recommended for the treatment of ADHD in children and adolescents.
Patients new to methylphenidate.
The recommended starting dose of Attora LA in patients who are currently not taking methylphenidate is 20 mg once daily.
When in the judgement of the clinician a lower initial dose is appropriate treatment may begin with Attora LA 10 mg.
Patients currently using methylphenidate.
Treatment may be continued with the same daily dose. If the patient was previously treated with an immediate release formulation, a switch to an appropriate recommended dose of Attora LA should be made (see Switching patient's treatment from methylphenidate immediate release tablets to Attora LA capsules).
Adults. Maximum daily doses.
Daily doses above 80 mg are not recommended for the treatment of ADHD in adults.
Patients new to methylphenidate.
The recommended starting dose of Attora LA in patients who are currently not taking methylphenidate is 20 mg once daily.
Patients currently using methylphenidate.
Treatment may be continued with the same daily dose. If the patient was previously treated with an immediate release formulation, a conversion to an appropriate recommended dose of Attora LA should be made (see Switching patient's treatment from Methylphenidate immediate release tablets to Attora LA capsules).
There are no differences recommended in dosing between male and female adult patients.
Use in elderly. Use of Attora LA in patients over 60 years of age has not been studies in controlled trials.
Switching patient's treatment from methylphenidate immediate release tablets to Attora LA capsules.
The recommended dose of Attora LA should be equal to the total daily dose of the immediate-release formulation not exceeding a total dose of 60 mg in children and 80 mg in adults. Examples involving switch from methylphenidate immediate-release tablets are provided in Table 1.
 For other methylphenidate regimens, clinical judgement should be used when selecting the starting dose.
For other methylphenidate regimens, clinical judgement should be used when selecting the starting dose.
Attora LA dosage may be adjusted at weekly intervals in 10 mg increments for children and in 20 mg increments for adults.
Administration.
Attora LA capsules should be administered orally once daily in the morning.
Attora LA may be swallowed as whole capsules or alternatively may be administered by sprinkling the capsule contents on a small amount of soft food (see specific instructions below). Attora LA capsules and/or their contents should not be crushed, chewed or divided.
The capsules may be administered with or without food. A high fat breakfast may slow the rate of absorption and therefore onset of action. Dosage should, therefore, be standardised in relation to food to ensure consistency of effect.
Attora LA administered as a single daily dose provides comparable overall exposure (AUC) of methylphenidate compared to the same total dose of immediate release tablets administered twice daily.
Sprinkling Attora LA capsule contents on food.
Carefully open the Attora LA capsule(s) and sprinkle the beads over applesauce. The food should not be warm because it could affect the modified-release properties of the formulation.
All of the mixture of drug and food should be immediately swallowed, unchewed. The drug and food mixture should not be stored for future use.4.3 Contraindications
Attora LA is contraindicated in patients with the following:
anxiety and tension states;
agitation;
a family history or diagnosis of Tourette's syndrome;
glaucoma;
hyperthyroidism;
pre-existing cardiovascular disorders including uncontrolled hypertension, angina pectoris, arterial occlusive disease especially coronary arteries; heart failure, haemodynamically significant congenital heart disease, cardiomyopathies, myocardial infarction, cardiac arrhythmia and channelopathies (disorders caused by the dysfunction of ion channels);
treatment with monoamine oxidase inhibitors, and also within a minimum of 14 days following discontinuation of a monoamine oxidase (MAO) inhibitor (hypertensive crises may result);
phaeochromocytoma;
known drug dependence or alcohol abuse;
severe depression, anorexia nervosa, psychotic symptoms or suicidal tendency, since methylphenidate might worsen these conditions;
known hypersensitivity to methylphenidate or to any component of the formulation.
4.4 Special Warnings and Precautions for Use
General.
Treatment with methylphenidate is not indicated in all cases of ADHD and should be considered only after detailed history taking and evaluation of the patient.
The decision to prescribe methylphenidate should depend on the physician's assessment of the chronicity and severity of the symptoms and in paediatric patients, the appropriateness to the child's age. Prescription should not depend solely on the presence of isolated behavioural characteristics. When the symptoms are associated with acute stress reactions, treatment with methylphenidate is usually not indicated.
Sudden death and pre-existing structural cardiac abnormalities or other serious heart problems.
It is essential that patients with pre-existing structural cardiac abnormalities or other serious heart problems being considered for treatment are assessed by a cardiologist before initiating treatment. On-going cardiological supervision should be maintained throughout treatment in these patients.
Children and adolescents.
Sudden death has been reported in association with CNS stimulant treatment at usual doses in children and adolescents with structural cardiac abnormalities or other serious heart problems.
Stimulant products, including methylphenidate, generally should not be used in patients with known serious structural cardiac abnormalities, cardiomyopathy, serious heart rhythm abnormalities, or other serious cardiac problems that may increase the risk of sudden death due to the sympathomimetic effects of a stimulant drug. Before initiating methylphenidate treatment, patients should be assessed for pre-existing cardiovascular disorders and a family history of sudden death and ventricular arrhythmia (see Section 4.2 Dose and Method of Administration).
Adults.
Sudden death, stroke, and myocardial infarction have been reported in adults taking stimulant drugs at usual doses for ADHD. Although the role of stimulants in these adult cases is also unknown, adults have a greater likelihood than children of having serious structural cardiac abnormalities, cardiomyopathy, serious heart rhythm abnormalities, coronary artery disease, or other serious cardiac problems. Adults with such abnormalities should also generally not be treated with stimulant drugs.
Cardiovascular conditions.
Attora LA is contraindicated in patients with severe hypertension. Methylphenidate increases heart rate and systolic and diastolic blood pressure. Therefore, caution is indicated in treating patients whose underlying medical conditions might be compromised by increases in blood pressure or heart rate, e.g. those with pre-existing hypertension. Severe cardiovascular disorders are contraindicated (see Section 4.3 Contraindications).
Methylphenidate should be used cautiously in patients with hypertension. Blood pressure should be monitored at appropriate intervals in all patients taking methylphenidate, especially in those with hypertension. Patients who develop symptoms suggestive of cardiac disease during methylphenidate treatment should undergo a prompt cardiac evaluation.
Children, adolescents, or adults who are being considered for treatment with stimulant medicine should have a careful history (including assessment for a family history of sudden death or ventricular arrhythmia) and physical exam to assess for the presence of cardiac disease, and should receive further cardiac evaluation if findings suggest such disease. Patients who develop symptoms such as exertional chest pain, unexplained syncope, or other symptoms suggestive of cardiac disease during stimulant treatment should undergo a prompt cardiac evaluation.
Misuse and cardiovascular events.
Misuse of stimulants of the central nervous system, including methylphenidate may be associated with sudden death and other serious cardiovascular adverse events.
Cerebrovascular.
Cerebrovascular conditions.
Patients with pre-existing central nervous system (CNS) abnormalities, e.g. cerebral aneurysm and/or other vascular abnormalities such as vasculitis or pre-existing stroke should not be treated with methylphenidate. Patients with additional risk factors (history of cardiovascular disease, concomitant medicine that elevates blood pressure) should be assessed regularly for neurological/psychiatric signs and symptoms after initiating treatment with methylphenidate (see Section 4.4 Special Warnings and Precautions for Use, Cardiovascular conditions; Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Psychiatric conditions.
Methylphenidate should not be used to treat severe depression or for the prevention or treatment of normal fatigue states. In psychotic patients administration of methylphenidate may exacerbate symptoms of behaviour disturbance and thought disorder.
Co-morbidity of psychiatric disorders in ADHD is common and should be taken into account when prescribing stimulant products. Prior to initiating treatment with methylphenidate, patients should be assessed for pre-existing psychiatric disorders and a family history of psychiatric disorders (see Section 4.2 Dose and Method of Administration).
Treatment of ADHD with stimulant products including methylphenidate should not be initiated in patients with acute psychosis, acute mania, or acute suicidality. These acute conditions should be treated and controlled before ADHD treatment is considered. Methylphenidate should not be used as treatment for severe depression of either exogenous or endogenous origin.
In the case of emergent psychiatric symptoms or exacerbation of pre-existing psychiatric symptoms, methylphenidate should not be given to patients unless the benefit outweighs the potential risk.
Psychotic symptoms.
Psychotic symptoms, including visual and tactile hallucinations or mania have been reported in patients administered usual prescribed doses of stimulant products, including methylphenidate (see Section 4.8. Adverse Effects (Undesirable Effects)). Physicians should consider treatment discontinuation if psychotic symptoms occur.
Bipolar illness.
Particular care should be taken in using stimulants to treat ADHD in patients with comorbid bipolar disorder because of concern for possible induction of a mixed/manic episode in such patients.
Aggressive behaviour.
Methylphenidate has been causally associated with aggression. Emergent aggressive behaviour or an exacerbation of baseline aggressive behaviour has been reported during stimulant therapy, including methylphenidate. Physicians should evaluate the need for adjustment of treatment regimen in patients experiencing these behavioural changes, bearing in mind that upwards or downwards titration may be appropriate. Treatment interruption can be considered.
Suicidal tendency.
Patients and caregivers of patients should be alerted about the need to monitor for clinical worsening, suicidal behaviour or thoughts or unusual changes in behaviour and to seek medical advice immediately if these symptoms appear. The physician should initiate appropriate treatment of the underlying psychiatric condition and consider a possible discontinuation or change in the ADHD treatment.
Motor and verbal tics.
CNS stimulants, including methylphenidate, have been associated with the onset or exacerbation of motor and verbal tics. Worsening of Tourette's syndrome has also been reported (see Section 4.8. Adverse Effects (Undesirable Effects)). Therefore, clinical evaluation for tics in patients should precede use of stimulant medicine. Family history should be assessed and clinical evaluation for tics or Tourette's syndrome in patients should precede use of methylphenidate for ADHD treatment. Attora LA is contraindicated in case of diagnosis or family history of Tourette's syndrome (see Section 4.3 Contraindications). Patients should be regularly monitored for the emergence or worsening of tics during treatment with methylphenidate.
Serotonin syndrome.
Serotonin syndrome has been reported following co-administration of methylphenidate with serotonergic drugs such as selective serotonin reuptake inhibitors (SSRIs) and serotonin-norepinephrine reuptake inhibitors (SNRIs). The concomitant use of methylphenidate and serotonergic drugs is not recommended as this may lead to the development of serotonin syndrome. The symptoms of serotonin syndrome may include mental status changes (e.g. agitation, hallucinations, delirium, and coma), autonomic instability (e.g. tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g. tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and/or gastrointestinal symptoms (e.g. nausea, vomiting, diarrhoea). Prompt recognition of these symptoms is important so that treatment with methylphenidate and serotonergic drugs can be immediately discontinued and appropriate treatment instituted (see Section 4.5 Interactions with Other Medicines and Other Forms of Interactions).
Paediatric use.
Methylphenidate should not be used in children under 6 years of age, since safety and efficacy in this age group have not been established. Medicines should be kept out of the reach of children.
Growth retardation.
Moderately reduced weight gain and slight growth retardation have been reported with the long term use of stimulants, including methylphenidate, in children (see Section 4.8. Adverse Effects (Undesirable Effects)). Growth should be monitored as clinically necessary during treatment with methylphenidate, and patients who are not growing or gaining height or weight as expected may need to have their treatment interrupted.
Careful follow up of weight and height in children aged 7 to 10 years who were randomised to either methylphenidate or non-medicine treatment groups over 14 months, as well as in naturalistic subgroups of newly methylphenidate-treated and non-medicine treated children over 36 months (to the ages of 10 to 13 years), suggests that consistently medicated children (i.e. treatment for 7 days per week throughout the year) have a temporary slowing in growth rate (on average, a total of about 2 cm less growth in height and 2.7 kg less growth in weight over 3 years), without evidence of growth rebound during this period of development.
Published data are inadequate to determine whether chronic use of amphetamines may cause similar suppression of growth, however, it is anticipated that they likely have this effect as well. Therefore, growth should be monitored during treatment with stimulants, and patients who are not growing or gaining height or weight as expected may need to have their treatment interrupted.
The retardation of growth referred to under Section 4.8. Adverse Effects (Undesirable Effects) is usually followed by catch-up growth when the medicine is discontinued. In order to minimise such complications, drug-free periods over weekends, school holidays and long vacations are advocated by some specialists.
Fatigue.
Methylphenidate should not be employed for the prevention or treatment of normal fatigue states.
Seizures.
There is some clinical evidence that methylphenidate may lower the convulsion threshold in patients with a history of seizures, with prior EEG abnormalities in the absence of seizures and, rarely, in the absence of a history of seizures and no prior EEG evidence of seizures. Safe concomitant use of anticonvulsants and methylphenidate has not been established. In the presence of seizures, the drug should be discontinued.
Acute angle closure glaucoma.
There have been reports of acute angle closure glaucoma associated with methylphenidate treatment. Although the mechanism is not clear, Ritalin-treated patients considered at risk for acute angle closure glaucoma (e.g. patients with significant hyperopia) should be evaluated by an ophthalmologist.
Increased intraocular pressure and glaucoma.
There have been reports of an elevation of intraocular pressure (IOP) and glaucoma (including open angle glaucoma and angle closure glaucoma) associated with methylphenidate treatment (see Section 4.8 Adverse Effects (Undesirable Effects)). Close monitoring of Ritalin-treated patients with a history of abnormally increased IOP or glaucoma is recommended.
Priapism.
Prolonged and painful erections, sometimes requiring surgical intervention, have been reported with methylphenidate products in both paediatric and adult patients. Priapism was not reported with drug initiation but developed after some time on the drug, often subsequent to an increase in dose. Priapism has also appeared during a period of drug withdrawal (drug holidays or discontinuation). Patients who develop abnormally sustained or frequent and painful erections should seek immediate medical attention.
Peripheral vasculopathy, including Raynaud's phenomenon.
Stimulants, including methylphenidate, used to treat ADHD are associated with peripheral vasculopathy, including Raynaud's phenomenon. Signs and symptoms are usually intermittent and mild; however, very rare sequelae include digital ulceration and/or soft tissue breakdown. Effects of peripheral vasculopathy, including Raynaud's phenomenon, have been observed in post marketing reports at different times and at therapeutic doses in all age groups throughout the course of treatment. Signs and symptoms generally improve after reduction in dose or discontinuation of drug. Careful observation for digital changes is necessary during treatment with ADHD stimulants. Further clinical evaluation (e.g. rheumatology referral) may be appropriate for certain patients.
Drug abuse and dependence.
Caution is called for in emotionally unstable patients, such as those with a history of drug or alcohol dependence, because they may increase the dosage on their own initiative. Chronic abuse can lead to marked tolerance and psychological dependence with varying degrees of abnormal behaviour. Frank psychotic episodes may occur, especially in response to parenteral abuse.
Clinical data indicate that children given methylphenidate are not more likely to abuse drugs as adolescents or adults. Methylphenidate abuse or dependence does not appear to be a problem in adolescents or adults who were treated with methylphenidate for ADHD as children.
Use with alcohol.
Alcohol may exacerbate the CNS adverse reactions of psychoactive drugs, including methylphenidate. Therefore, it is advisable for patients to abstain from alcohol during treatment.
Withdrawal.
Careful supervision is required during drug withdrawal, since this may unmask depression as well as the effects of chronic over-activity. Some patients may require long-term follow-up.
Haematological effects.
Data on safety and efficacy of long-term use of methylphenidate are not complete. Patients requiring long-term therapy should be carefully monitored and periodic complete blood counts, differential and platelet counts are advisable during prolonged therapy. In the event of haematological disorders appropriate medical intervention should be considered (see Section 4.8. Adverse Effects (Undesirable Effects)).
Use in the elderly.
See Section 4.2 Dose and Method of Administration.
Effects on laboratory tests.
Methylphenidate may induce false positive laboratory tests for amphetamines, particularly with immunoassays screen test.4.5 Interactions with Other Medicines and Other Forms of Interactions
Anti-hypertensive drugs.
Methylphenidate may decrease the effectiveness of drugs used to treat hypertension.
Use with drugs that elevate blood pressure.
Methylphenidate should be used with caution in patients being treated with drugs that elevate blood pressure due to the risk of severe hypertension (see Section 4.4. Special Warnings and Precautions for Use, Cerebrovascular conditions).
Because of possible hypertensive crisis, Attora LA is contraindicated in patients being treated (currently or within the preceding 2 weeks) with non-selective, irreversible MAO inhibitors (see Section 4.3 Contraindications).
Use with anaesthetics.
There is a risk of sudden blood pressure and heart rate increase during surgery. If surgery is planned, Attora LA should not be taken on the day of surgery.
Use with centrally acting alpha-2 agonists (e.g. clonidine).
Serious adverse events including sudden death have been reported in concomitant use with clonidine, although no causality for the combination has been established.
Use with dopaminergic drugs.
As an inhibitor of dopamine reuptake, methylphenidate may be associated with pharmacodynamic interactions when co-administered with direct and indirect dopamine agonists (including DOPA and tricyclic antidepressants) as well as dopamine antagonists (antipsychotics, e.g. haloperidol).
Concomitant use of methylphenidate with antipsychotics is not recommended due to its counteracting mechanism of action. If upon medical assessment the combination is deemed necessary, monitoring for extrapyramidal symptoms (EPS) is recommended, as the concomitant use of methylphenidate with antipsychotics may increase the risk of EPS when there is a change (increase or decrease) in dosage of either or both medications.
Use with serotonergic drugs.
The concomitant use of methylphenidate and serotonergic drugs is not recommended as this may lead to the development of serotonin syndrome (see Section 4.4. Special Warnings and Precautions for Use). Methylphenidate has been shown to increase extracellular serotonin and noradrenaline and appears to have weak potency in binding serotonin transporter.
Pharmacokinetic interactions.
Methylphenidate is not metabolized by cytochrome P450 to a clinically relevant extent. Inducers or inhibitors of cytochrome P450 are not expected to have any relevant impact on methylphenidate pharmacokinetics. Conversely, the d- and l-enantiomers of methylphenidate did not relevantly inhibit in vitro cytochrome P450 1A2, 2C8, 2C9, 2C19, 2D6, 2E1 and 3A.
Methylphenidate coadministration did not increase plasma concentrations of the CYP2D6 substrate desipramine.
Case reports have shown that methylphenidate may inhibit the metabolism of coumarin anticoagulants, anticonvulsants (phenobarbital, primidone, phenytoin), phenylbutazone and tricyclic antidepressants (imipramine, desipramine), but pharmacokinetic interactions were not confirmed when explored at higher sample sizes. Reduction in the dosage of these drugs may be required when they are given concomitantly with methylphenidate.
Serious adverse events have been reported in concomitant use with clonidine, although no causality for the combination has been established. The safety of using methylphenidate in combination with clonidine or other centrally acting alpha-2 agonists has not been systematically evaluated.
Other specific drug-drug interaction studies with methylphenidate have not been performed in vivo.4.6 Fertility, Pregnancy and Lactation
Effect on fertility.
No human data on the effect of methylphenidate on fertility are available.
Women of child-bearing potential.
Methylphenidate should not be prescribed for women of childbearing age unless, in the opinion of the physician, the potential benefits outweigh the possible risks (see Use in pregnancy).
(Category D)
The safety of methylphenidate for use during human pregnancy has not been established. Data from a cohort study of in total approximately 3,400 pregnancies exposed in the first trimester do not suggest an increased risk of overall birth defects. There was a small increased occurrence of cardiac malformations in women who receive methylphenidate during the first trimester of pregnancy, compared with non-exposed pregnancies. Methylphenidate should not be prescribed for pregnant women unless, in the opinion of the physician, the potential benefits outweigh the possible risks.
As a general rule no drugs should be taken during the first 3 months of pregnancy, and the benefits and risks of taking drugs should be carefully considered throughout the whole of the pregnancy.
Case reports showed that methylphenidate was distributed into breast milk reaching a milk-to-plasma ratio of approximately 2.5. For safety reasons, mothers taking methylphenidate should refrain from breast-feeding their infants. A decision should be made by the prescriber whether the mother must abstain from breast-feeding or abstain from methylphenidate therapy, taking into account the benefit of breast-feeding to the child and the benefit of therapy to the mother.4.7 Effects on Ability to Drive and Use Machines
Methylphenidate may cause dizziness, drowsiness, blurred vision, hallucinations or other CNS side effects (see Section 4.8 Adverse Effects (Undesirable Effects)). Patients experiencing such side effects should refrain from driving, operating machinery, or engaging in other potentially hazardous activities.
4.8 Adverse Effects (Undesirable Effects)
Adverse effects reported during clinical trials.
Children.
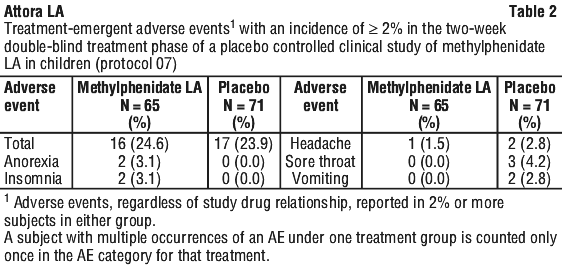
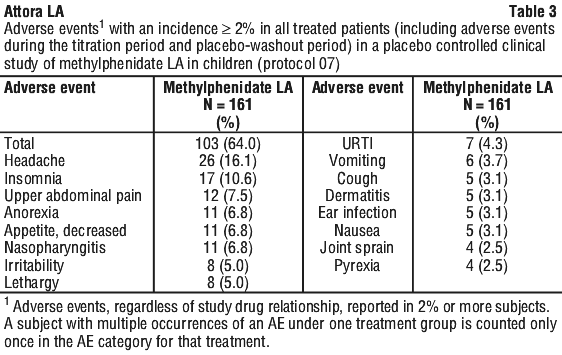
A randomised, double-blind, placebo-controlled parallel group clinical study (Protocol 07) was conducted to evaluate the efficacy and safety of methylphenidate modified release (LA) capsules in children aged 6 - 12 years with ADHD. In the double-blind treatment phase of this study, methylphenidate modified release was administered once daily for up to two weeks at individually titrated doses in the 10 - 40 mg range. The incidence of treatment-emergent adverse events reported during the double-blind treatment phase is summarised in Table 2. Adverse events reported with an incidence ≥ 2% in all treated subjects in Protocol 07 (including adverse events during the titration period and placebo-washout period) are included in Table 3.
Adults.
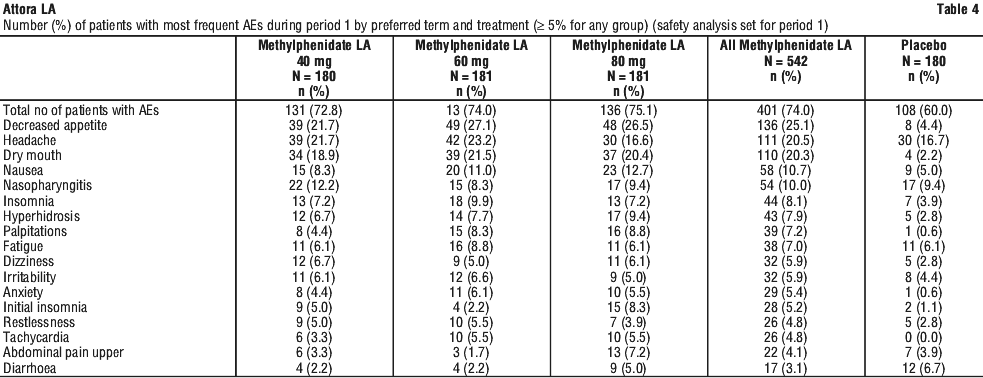
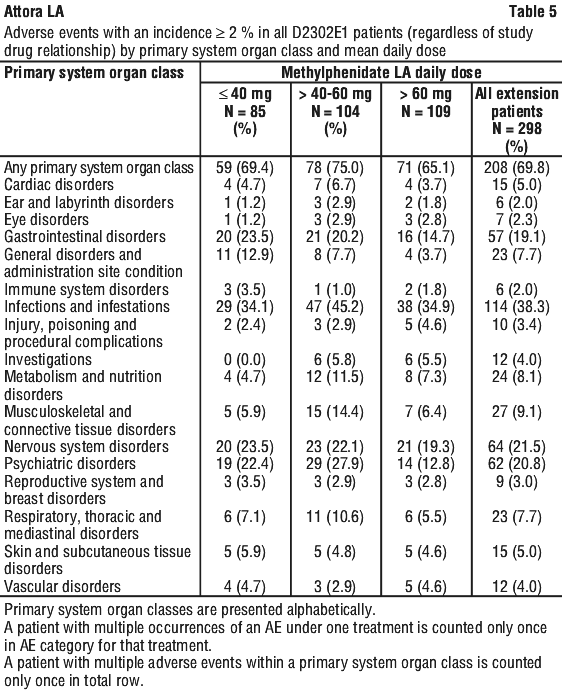
Methylphenidate modified release capsules was evaluated in a randomized, double-blind, placebo-controlled, multicentre core study (D2302) in the treatment of 725 adult patients (395 male and 330 female) diagnosed with ADHD. Maintenance of effect of methylphenidate modified release capsules was evaluated in a 6-month maintenance study (D2302E1) in 298 patients. The incidence of adverse events reported during the core study and extension studies are summarised in Table 4 and Table 5 respectively.
 The AE profile seen in the maintenance period was similar to that observed in the core study. No unexpected SAEs or AEs were observed in this extension study and the commonly observed AEs were expected and driven by the pharmacologic activity.
The AE profile seen in the maintenance period was similar to that observed in the core study. No unexpected SAEs or AEs were observed in this extension study and the commonly observed AEs were expected and driven by the pharmacologic activity.
The most frequently affected primary SOCs (≥ 10% in all extension patients) starting in the extension study were infections and infestations, nervous system disorders, psychiatric disorders, and gastrointestinal disorders. There was no evidence of toxicity to any organ system. The most commonly reported individual AEs (≥ 5.0% in any dose group) during the extension study were nasopharyngitis (19.1%), headache (14.1%), decreased appetite (7.7%), dry mouth (6.7%) and nausea (5.0%).
Post-marketing experience.
Nervousness and insomnia are very common adverse reactions which occur at the beginning of methylphenidate treatment and are usually controlled by reducing the dosage and omitting the drug in the afternoon or evening.
Loss of appetite is very common but usually transient. Abdominal pain, insomnia and tachycardia are common, usually at the beginning of treatment and may be alleviated by concomitant food intake.
Tabulated summary of adverse drug reactions. Adverse drug reactions listed in Table 6 are listed by MedDRA system organ class. Within each system organ class, the adverse drug reactions are ranked by frequency, with the most frequent reactions first. Within each frequency grouping, adverse drug reactions are presented in order of decreasing seriousness. In addition, the corresponding frequency category for each adverse drug reaction is based on the following convention (CIOMS III): very common ≥ 10%; common ≥ 1% to < 10%; uncommon ≥ 0.1% to < 1%; rare ≥ 0.01% to < 0.1%; very rare < 0.01%.
 Very rare reports of poorly documented neuroleptic malignant syndrome (NMS) have been received. In most of these reports, patients were also receiving other medications. It is uncertain what role methylphenidate played in these cases.
Very rare reports of poorly documented neuroleptic malignant syndrome (NMS) have been received. In most of these reports, patients were also receiving other medications. It is uncertain what role methylphenidate played in these cases.
Adverse events reported since market introduction in patients taking methylphenidate include suicide, suicide attempt and suicidal ideation. No causal relationship between methylphenidate and these events has been established.
Adverse drug reactions from spontaneous reports and literature cases (frequency not known). The following adverse drug reactions have been derived from post-marketing experience with methylphenidate via spontaneous case reports and literature cases. Because these reactions are reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency which is therefore categorized as not known. Adverse drug reactions are listed according to system organ classes in MedDRA. Within each system organ class, ADRs are presented in order of decreasing seriousness.
 Additional adverse reactions reported with other methylphenidate-containing products. The list below shows adverse reactions not listed for methylphenidate that have been reported with other methylphenidate-containing products based on clinical trials data and post-market spontaneous reports.
Additional adverse reactions reported with other methylphenidate-containing products. The list below shows adverse reactions not listed for methylphenidate that have been reported with other methylphenidate-containing products based on clinical trials data and post-market spontaneous reports.
Immune system disorders.
Hypersensitivity reactions such as auricular swelling.
Renal and urinary disorders.
Haematuria, incontinence.
General disorders and administration site conditions.
Sudden cardiac death.
Investigations.
Cardiac murmur.
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
Symptoms.
Signs and symptoms of acute overdosage, mainly due to over-stimulation of the central nervous system and from excessive sympathomimetic effects, may include: vomiting, agitation, tremors, hyperreflexia, muscle twitching, convulsions (may be followed by coma), euphoria, confusion, hallucinations, delirium, sweating, flushing, headache, hyperpyrexia, tachycardia, palpitation, cardiac arrhythmias, hypertension, mydriasis, dryness of mucous membranes and rhabdomyolysis.
Treatment.
When treating an overdose, practitioners should bear in mind that a second release of methylphenidate from Attora LA capsules occurs at approximately four hours after administration of the capsule.
Treatment consists of appropriate supportive measures and symptomatic treatment of life-threatening events, e.g. hypertensive crisis, cardiac arrhythmias, convulsions. For the most current guidance for treatment of symptoms of overdose, the practitioner should consult the Poisons Information Centre on 13 11 26 or current toxicological publication.
The patient must be protected against self-injury and against external stimuli that would aggravate overstimulation already present. If the signs and symptoms are not too severe and the patient is conscious, further absorption may be limited by administration of activated charcoal. In cases of marked agitation, intravenous doses of diazepam or haloperidol should be given. Hypertension may be controlled by alpha-adrenergic blocking agents or intravenous sodium nitroprusside.
Intensive care must be provided to maintain adequate circulation and respiratory exchange; external cooling procedures may be required for hyperpyrexia.
The efficacy of peritoneal dialysis or extracorporeal haemodialysis for methylphenidate overdosage has not been established. Clinical experience with overdose is limited. Patients who have received doses higher than those recommended should be carefully monitored. In the event of overdose leading to clinically significant hypocalcaemia, reversal may be achieved with supplemental oral calcium and/or infusion of calcium gluconate.5 Pharmacological Properties
Pharmacodynamic pharmacotherapeutic group: psychostimulants.
ATC code: N06BA04.
5.1 Pharmacodynamic Properties
Methylphenidate is a racemate consisting of a 1:1 mixture of d-threo methylphenidate (d-MPH) and l-threo methylphenidate (l-MPH).
Mechanism of action.
Methylphenidate is a central nervous system (CNS) stimulant. Its mode of action in humans is not completely understood but methylphenidate presumably exerts its stimulant effect by an inhibition of dopamine and norepinephrine reuptake into presynaptic neurons and thereby increasing these neurotransmitters in the extraneuronal space. There is neither specific evidence which clearly establishes the mechanism whereby methylphenidate produces its mental and behavioural effects in children, nor conclusive evidence as to how these effects relate to the condition of the central nervous system.
The l-enantiomer is thought to be pharmacologically inactive.
Repeated oral administration of methylphenidate to young rats was associated with decreased spontaneous locomotor activity at systemic exposures (plasma AUC) about 3-fold that at the maximum clinical dose, due to an exaggerated pharmacological activity of methylphenidate. A deficit in the acquisition of a specific learning task was also observed, only in females, at systemic exposures (plasma AUC) 8-fold that at the maximum clinical dose. The clinical relevance of these findings is unknown.
The effect of treatment with 40 mg dexmethylphenidate hydrochloride, the pharmacologically active d-enantiomer of methylphenidate, on QT/QTc interval was evaluated in a study in 75 healthy volunteers. The maximum mean prolongation of QTcF intervals was < 5 ms, and the upper limit of the 90% confidence interval was below 10 ms for all time matched comparisons versus placebo. This was below the threshold of clinical concern and no exposure response relationship was evident.
Clinical trials.
ADHD in children.
Methylphenidate modified release LA capsules was demonstrated to be effective in the treatment of ADHD in two placebo-controlled clinical studies in children aged 6 - 12 years with a DSM-IV diagnosis of ADHD (n = 195 exposed to study medicine).
The results of the first study (Protocol 02), a randomised, double-blind, placebo controlled, crossover clinical study conducted in a laboratory classroom setting, demonstrated that a single dose of 20 mg Methylphenidate LA had a rapid onset of effect and was statistically superior to placebo in improving classroom behaviour and cognitive responses. After administration of Methylphenidate LA, the improvement relative to placebo was statistically significant during both the morning (0 - 4 hours) and the afternoon (4 - 9 hours).
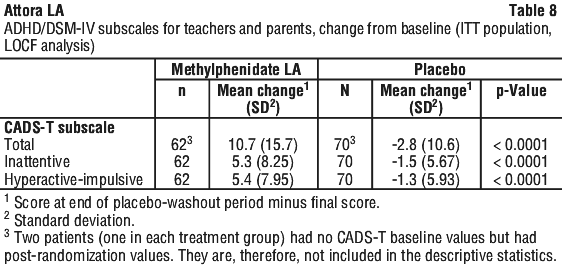
The second study (Protocol 07) was a randomised, double-blind, placebo controlled, parallel group clinical study conducted in normal school and home settings, in which Methylphenidate LA was administered once daily at individually titrated doses in the range of 10 - 40 mg/day for up to two weeks. The primary efficacy variable was the change from baseline to final score in the Connors ADHD/DSM-IV Scale for Teachers (CADS-T). CADS-T assesses symptoms of hyperactivity and inattention. The results of the analysis demonstrate that Methylphenidate LA was statistically superior to placebo (p < 0.0001). The efficacy of Methylphenidate LA in controlling symptoms of ADHD was consistently reflected in the assessments of teachers, parents and investigators. The results of the primary efficacy analysis is summarised in Table 8.
ADHD in adults.
Methylphenidate modified release LA capsules was evaluated in a randomized, double-blind, placebo-controlled, multicentre study in the treatment of 725 adult patients (395 male and 330 female) diagnosed with ADHD according to DSM-IV ADHD criteria. The study was designed to:
1) Confirm the clinically effective and safe dose range of Methylphenidate LA for adults (18 to 60 years old) in a 9-week, double-blind, randomized, placebo-controlled, parallel group period (Period 1) consisting of a 3-week titration stage followed by a 6-week fixed dose stage (40, 60, 80 mg/day or placebo). Subsequently patients were re-titrated to their optimal dose of Methylphenidate LA (40, 60 or 80 mg/day) over a 5 week period (Period 2).
2) Evaluate the maintenance of effect of Methylphenidate LA in adults with ADHD in a 6 month, double-blind, randomized, withdrawal study (period 3).
Efficacy was assessed using the DSM-IV ADHD rating scale (DSM-IV ADHD RS) for symptomatic control and Sheehan Disability Score (SDS) for functional improvement as change in respective total scores from baseline to the end of the first period. All dose levels of Methylphenidate LA showed significantly greater symptom control (p < 0.0001 for all dose levels) compared to placebo as measured by a reduction in DSM-IV ADHD RS total score. All doses of Methylphenidate LA showed significantly greater functional improvement (p = 0.0003 at 40 mg, p = 0.0176 at 60 mg, p < 0.0001 at 80 mg) compared to placebo as measured by reduction in SDS total score (see Table 9).
 Significant clinical efficacy was demonstrated in all three Methylphenidate LA dose levels using physician rated scales [Clinical Global Impression- Improvement (CGI-I) and Clinical Global Improvement-Severity (CGI-S)], self-rated scales [Adult Self-Rating Scale (ASRS)] and observer- rated scales [Conners' Adult ADHD Rating Scale Observer Short Version (CAARS O:S)]. The results were consistently in favour of Methylphenidate LA over placebo across all assessments in period 1.
Significant clinical efficacy was demonstrated in all three Methylphenidate LA dose levels using physician rated scales [Clinical Global Impression- Improvement (CGI-I) and Clinical Global Improvement-Severity (CGI-S)], self-rated scales [Adult Self-Rating Scale (ASRS)] and observer- rated scales [Conners' Adult ADHD Rating Scale Observer Short Version (CAARS O:S)]. The results were consistently in favour of Methylphenidate LA over placebo across all assessments in period 1.
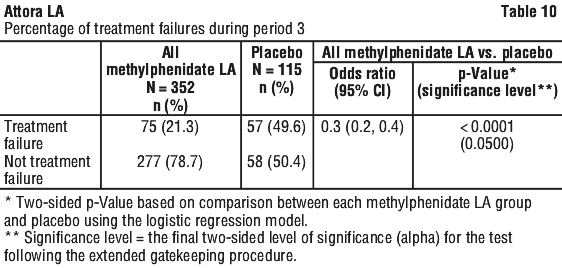
Maintenance of effect of Methylphenidate LA was evaluated by measuring the percentage of treatment failure in Methylphenidate LA compared to the placebo group at the end of a 6-month maintenance period (see Table 10). Completion rates varied across the treatment groups, with the lowest rate in the placebo group (34.1%) and rates between 47.7% and 58.3% across the Methylphenidate LA dose group. Once the Methylphenidate LA dose was optimized in Period 2, approximately 79 % of patients continued to maintain disease control for a period of at least 6 months compared with approximately 50% given placebo (p < 0.0001 vs. placebo). An odds ratio of 0.3 suggested that patients treated with placebo had a 3 times higher chance of becoming a treatment failure compared to Methylphenidate LA.
 Patients who entered Period 3 had completed a total of between 5-14 weeks of Methylphenidate LA treatment in Periods 1 and 2. Patients then assigned to placebo in Period 3 did not experience increased signs of withdrawal and rebound compared to patients who continued on Methylphenidate LA treatment. The study performed in adults did not suggest any difference in efficacy or safety amongst gender subgroups (see Section 4.2 Dose and Method of Administration.
Patients who entered Period 3 had completed a total of between 5-14 weeks of Methylphenidate LA treatment in Periods 1 and 2. Patients then assigned to placebo in Period 3 did not experience increased signs of withdrawal and rebound compared to patients who continued on Methylphenidate LA treatment. The study performed in adults did not suggest any difference in efficacy or safety amongst gender subgroups (see Section 4.2 Dose and Method of Administration.
Efficacy was maintained or improved in the majority of subjects who elected to continue open-label treatment with Methylphenidate LA for up a further 26 weeks. Symptomatic improvement and a reduction in functional impairment was maintained or improved throughout the 26 week extension study period in the patients who elected to continue open label treatment with Methylphenidate LA.
5.2 Pharmacokinetic Properties
Absorption.
Following oral administration of methylphenidate modified release LA capsule to children diagnosed with Attention-Deficit Hyperactivity Disorder (ADHD) and adults, methylphenidate is rapidly absorbed and produces a bimodal plasma concentration-time profile (i.e. two distinct peaks approximately four hours apart). The relative bioavailability methylphenidate LA capsules administered once daily is comparable to the same total dose of immediate release methylphenidate HCl tablets administered twice daily in children and in adults.
The fluctuations between peak and trough plasma methylphenidate concentrations are smaller for methylphenidate LA capsules administered once daily compared to methylphenidate immediate release tablets administered twice daily.
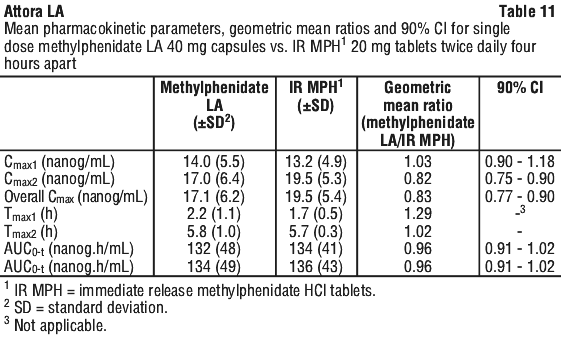
The mean pharmacokinetic parameters for methylphenidate LA 40 mg capsules administered as a single dose once daily compared to methylphenidate HCl 20 mg immediate-release tablets administered twice daily four hours apart are summarised in Table 11.
Food effects.
Attora LA may be administered with or without food. There were no significant differences in AUC when methylphenidate LA was administered with either a high fat breakfast or applesauce, compared to administration in the fasting condition, although a high fat meal and applesauce reduced Cmax2 by 25% and 13% respectively. There is no evidence of dose dumping in the presence or absence of food.
For patients unable to swallow the capsule, the contents may be sprinkled on applesauce and administered (see Section 4.2 Dose and Method of Administration).
Distribution.
In the blood, methylphenidate and its metabolites become distributed in the plasma (57%) and the erythrocytes (43%). Methylphenidate and its metabolites have low plasma protein-binding (approximately 15%). The apparent volume of distribution (Vd) has been calculated at 13.1 L/kg after an oral dose. The volume of distribution was 2.65 ± 1.11 L/kg for d-MPH and 1.80 ± 0.91 L/kg for l-MPH, following intravenous administration of 10 mg MPH.
Methylphenidate excretion into breast milk has been noted in two case reports where the calculated relative infant dose was ≤ 0.2 % of the weight adjusted maternal dose (see Section 4.6 Fertility, Pregnancy and Lactation, Use in lactation). Adverse events were not noted in either infant (6 months and 11 months of age).
Metabolism.
Biotransformation of methylphenidate, primarily by the carboxyl esterase CES1A1, is rapid and extensive. Peak plasma concentrations of the main, deesterified, metabolite, α-phenyl-2-piperidine acetic-acid (ritalinic acid), are attained about 2 hours after administration and are 30 to 50 times higher than those of the unchanged substance. The half-life of α-phenyl-2-piperidine acetic acid is about twice that of methylphenidate. Only small amounts of hydroxylated metabolites (e.g. hydroxymethylphenidate and hydroxyritalinic acid) are detectable. Therapeutic activity seems to be principally due to the parent compound.
Excretion.
Methylphenidate is eliminated from the plasma with a mean half-life of 2 to 3 hours, and the calculated mean systemic clearance is 4 to 10 L/h/kg after an oral dose. The systemic clearance is 0.40±0.12 L/h/kg for d-MPH and 0.73±0.28 L/h/kg for l-MPH. Within 48 to 96 hours, 78 to 97% of the dose administered is excreted in the urine and 1 to 3% in the faeces in the form of metabolites. Unchanged methylphenidate appears in the urine only in small quantities (< 1%). Most of the dose is excreted in the urine as α-phenyl-2-piperidine acetic acid (60-86%).
Special populations.
Effect of age.
There are no apparent differences in the pharmacokinetic behaviour of methylphenidate in hyperactive children and healthy adult volunteers.
Patients with renal impairment.
Elimination data from patients with normal renal function suggest that renal excretion of unchanged methylphenidate would hardly be diminished in the presence of impaired renal function. However, renal excretion of the metabolite α-phenyl-2-piperidine acetic acid may be reduced.
5.3 Preclinical Safety Data
Fertility.
Methylphenidate did not impair fertility in male or female mice that were fed diets containing the drug in an 18 week Continuous Breeding study. The study was conducted at doses up to 160 mg/kg/day, approximately 11-fold the highest recommended human dose of methylphenidate on a mg/m2 basis.
Reproductive animal toxicity.
Adequate animal reproduction studies to establish safe use of methylphenidate during pregnancy have not been conducted. Oral administration of methylphenidate to rabbits during the period of organogenesis has produced teratogenic effects at systemic exposures (plasma AUC) approximately 3 times clinical exposure at the maximum recommended human dose. The exposure at the no-effect dose was less than human exposure. In rats, teratogenic effects were not seen at systemic exposures (plasma AUC) approximately 12 times clinical exposure at the maximum recommended human dose.
Genotoxicity.
Methylphenidate was not mutagenic in assays in vitro (Ames reverse mutation assay and the mouse lymphoma cell forward mutation assay). Methylphenidate showed evidence of a weak clastogenic response in vitro (Chinese Hamster Ovary cells) but was negative in vivo (mouse bone marrow micronucleus assay).
Carcinogenicity.
In a lifetime carcinogenicity study carried out in B6C3F1 mice, methylphenidate caused an increase in hepatocellular adenomas and, in males only, an increase in hepatoblastomas at a daily dose of approximately 60 mg/kg/day. This dose is approximately 4 times the maximum recommended human dose of methylphenidate on a mg/m2 basis. Hepatoblastoma is a relatively rare rodent malignant tumour type. The mouse strain used is sensitive to the development of hepatic tumours, and the significance of these results to humans is unknown.
Methylphenidate did not cause any increases in tumours in a lifetime carcinogenicity study carried out in F344 rats; the highest dose used was approximately 50 mg/kg/day, which is approximately 7 times the maximum recommended human dose of methylphenidate on a mg/m2 basis.
In a 24-week carcinogenicity study in the transgenic mouse strain p53+/-, there was no evidence of carcinogenicity. Male and female mice were fed diets containing the same concentration of methylphenidate as in the lifetime carcinogenicity study; approximately 60 and 74 mg/kg/day of methylphenidate, respectively, which is approximately 4 and 5 times the maximum recommended human dose of methylphenidate on a mg/m2 basis, respectively.
Comment.
The US Food and Drug Administration examined data from the Surveillance, Epidemiology and End Results (SEER) database for the years 1973 to 1991 and found that the estimated incidence of hepatoblastoma in the general population was not greater than 1 in 10 million person-years.
A total of 174 cases of hepatoblastoma were reported by the SEER for the period 1973 to 1995. The age-adjusted incidence rate is very low (IR=0.0382 per 100,000 person-years). The majority of cases (149 out of 174) were diagnosed among the age group 0 to 4 years old, which is in accordance with the natural history of the disease. For the age group 5 to 24 years old the rates of hepatoblastoma are very low with 14 cases reported. For the 0 to 4 years old age group, incidence rates of hepatoblastoma have risen slowly, ranging from 0.3032 per 100,000 in 1973 to 0.4889 per 100,000 in 1995. On the basis of experience since marketing methylphenidate immediate release tablets, there is no evidence that the incidence is higher in patients receiving methylphenidate.6 Pharmaceutical Particulars
6.1 List of Excipients
Attora LA capsules contain microcrystalline cellulose, hypromellose, methacrylic acid copolymer, purified talc, triethyl citrate, ethylcellulose, hyprolose, gelatin, titanium dioxide, iron oxide yellow (only for 10 mg, 30 mg, 40 mg and 60 mg capsules) and Capsugel ink 10A2 Black (ARTG PI No. 109521).
6.2 Incompatibilities
Incompatibilities were either not assessed or not identified as part of the registration of this medicine.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
6.4 Special Precautions for Storage
Attora LA capsules.
Store below 25°C.
6.5 Nature and Contents of Container
Attora LA capsules.
HDPE bottles with child resistant PP caps, containing 30 capsules.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
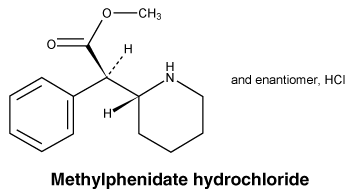
Chemical structure.
Active ingredient: Methylphenidate hydrochloride.
Chemical names: Methyl (2RS)-phenyl[(2RS)-piperidin-2-yl] acetate hydrochloride.
Molecular formula: C14H16NO2 . HCl.
Molecular weight: 269.8.
CAS number.
298-59-9.7 Medicine Schedule (Poisons Standard)
Controlled Drug (S8).
Summary Table of Changes
