What is in this leaflet
This leaflet answers some common questions about AURORIX tablets. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking AURORIX tablets against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine.
You may need to read it again.
What AURORIX is used for
The name of your medicine is AURORIX. It contains the active ingredient called moclobemide.
AURORIX belongs to a group of medicines called antidepressants. Antidepressants are used to treat depression and work on the central nervous system. They are thought to work by their action on brain chemicals called amines, which are involved in controlling mood.
There are many different types of medicines used to treat depression. AURORIX belongs to a group of medicines called reversible inhibitors of monoamine oxidase A.
Your doctor, however, may have prescribed AURORIX for another purpose.
Ask your doctor if you have any questions about why AURORIX has been prescribed for you.
This medicine is available only with a doctor's prescription.
Do not give AURORIX to children or adolescents under 18 years of age.
The safety and effectiveness of AURORIX in this age group has not been established.
Before you take AURORIX
When you must not take it
Do not take AURORIX if you have ever had an allergic reaction to AURORIX or any of the ingredients listed in the 'Ingredients' section of this leaflet.
Do not take AURORIX if you are suffering from severe confusion.
Do not take AURORIX if you are taking:
- clomipramine
- selegiline
- bupropion
- triptans (a family of medicines commonly used to treat migraines e.g. zolmitriptan)
- pethidine
- tramadol
- dextromethorphan (often found in cough and cold medicines)
- linezolid.
Do not take AURORIX if you are taking other medications known as selective serotonin reuptake inhibitors or tricyclic antidepressants.
Taking AURORIX with these medicines may cause a serious reaction called serotonin syndrome. This can cause a sudden increase in body temperature, high blood pressure and convulsions.
Do not take AURORIX after the expiry date (EXP) printed on the pack.
It may have no effect at all or, worse, an entirely unexpected effect if you take it after the expiry date.
Do not take AURORIX if the packaging is torn or shows signs of tampering or if the tablets appear damaged in some way.
If it has expired or is damaged, return the product to your pharmacist for disposal.
If you are not sure if you should be taking AURORIX, talk to your doctor.
Before you start to take it
Tell your doctor if you are allergic to any other medicines, foods, dyes or preservatives.
Tell your doctor if you have any other health problems including:
- liver disease
- high blood pressure
- a personal history or family history of bipolar disorder
- mental illness other than depression, including schizophrenia, agitation and excitation
- thyrotoxicosis (a condition of excessive thyroid hormones)
- phaeochromocytoma (a rare tumour of adrenal gland)
- rare hereditary problems of galactose intolerance, the Lapp lactose deficiency or glucose-galactose malabsorption.
Tell your doctor if you are pregnant or plan to become pregnant.
Tell your doctor if you are breastfeeding or wish to breastfeed.
Your doctor will discuss the risks and benefits of using AURORIX when pregnant and while breastfeeding.
If you have not told your doctor about any of the above, tell them before you start taking AURORIX.
Taking other medicines
Tell your doctor if you are taking any other medicines, including any that you have bought from a pharmacy, supermarket or health food shop.
In addition to the medications that are contraindicated with AURORIX, some other commonly used medicines that may interfere with AURORIX are:
- cimetidine
- dextropropoxyphene
- proton pump inhibitors
- serotonin agonists (e.g. buspirone, sumatriptan)
- St. John’s Wort (hypericum) - containing phytotherapeutic products.
- opiates e.g. morphine, fentanyl and codeine
- adrenergics
- sibutramine
Other antidepressant medicines that may interfere with AURORIX include: fluoxetine, fluvoxamine, paroxetine, sertraline, amitriptyline and nortriptyline, trimipramine and maprotiline, venlafaxine, clomipramine, citalopram and paroxetine.
Moclobemide may cause an additional drop in blood pressure if you are taking metoprolol.
Your doctor or pharmacist has a complete list of medicines to avoid while taking AURORIX.
How to take AURORIX
Take AURORIX exactly as your doctor has prescribed.
How much to take
Your doctor will tell you how many AURORIX tablets to take each day.
The usual dose is between 300 mg and 600 mg per day.
How to take AURORIX
Tablets should be swallowed whole with a glass of water after meals.
You should follow your doctor's instructions carefully if changing from one antidepressant to another and report any unexpected effects if they occur.
When to take AURORIX
AURORIX should be taken morning and evening at the end of your meal.
How long to take AURORIX
For depression, the length of treatment will depend on how quickly your symptoms improve. Most antidepressants take time to work so don't be discouraged if you don't feel better right away. Some of your symptoms may improve in 1 or 2 weeks but, it can take up to 4 or 6 weeks to feel any real improvement. Even when you feel well, you will usually have to take AURORIX for several months or even longer to make sure the benefits will last. Continue taking it until your doctor tells you to stop.
If you forget to take AURORIX
If it is almost time for your next dose, skip the dose you missed and take your next dose when you are meant to. Otherwise, take it as soon as you remember then go back to taking it as soon as you would normally.
Do not double a dose to make up for one you have missed.
If you take too much (overdose)
Immediately telephone your doctor or the Poisons Information Centre on telephone 13 11 26 for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much AURORIX. Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Keep telephone numbers for these places handy.
Some signs and symptoms of overdose include nausea, vomiting, drowsiness, slurred speech, reduced reflexes and agitation.
While you are taking AURORIX
Things you must do
Tell all doctors, dentists, and pharmacists who are treating you that you are taking AURORIX.
Do not take any other medicines whether they require a prescription or not without first telling your doctor.
Tell your doctor if you become pregnant while taking AURORIX.
Tell your doctor if, for any reason, you have not taken your medicine exactly as prescribed.
Otherwise, your doctor may think that it was not effective and change your treatment unnecessarily.
Tell your doctor if you feel the tablets are not helping your condition.
If you are being treated for depression, tell your doctor immediately if you feel your condition has worsened or if you are experiencing suicidal thoughts.
Be sure to discuss with your doctor any problems you may have and how you feel. This will help your doctor to determine the best treatment for you.
Be sure to keep all of your appointments with your doctor so that your progress can be checked.
Things you must not do
Do not stop taking AURORIX or lower the dose without first checking with your doctor.
Do not let yourself run out of medicine over the weekend or on holidays.
Do not give this medicine to anyone else even if their symptoms seem similar to yours.
Do not use AURORIX to treat other complaints unless your doctor says to.
Things to be careful of
Be careful driving or operating machinery until you know how AURORIX affects you.
AURORIX causes dizziness in some people at first.
Although drinking alcohol is unlikely to affect your response to AURORIX, your doctor may suggest avoiding alcohol while you are being treated for depression.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking AURORIX.
AURORIX helps most people with depression, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
In the first week or two you may experience the following:
- sleep disturbances
- dizziness
- nausea
- headache
- dry mouth
Occasionally, the symptoms of depression may include thoughts of suicide or self-harm. These symptoms may continue or get worse during the first one to two months of treatment until the full antidepressant effect of the medicine becomes apparent. This is more likely to occur if you are a young adult i.e. under 24 years of age.
Contact your doctor or a mental health professional right away, or go to Accident and Emergency at your nearest hospital for treatment if you or someone you know is demonstrating any of the following warning signs of suicide while taking AURORIX:
- thoughts or talk of death or suicide
- thoughts or talk of self-harm or harm to others
- any recent attempts of self-harm
- increase in aggressive behaviour, irritability or agitation
- worsening of depression
- insomnia, nervousness, jitteriness
- mania or hypomania (or onset of early symptoms).
All thoughts or talk of suicide or violence must be taken seriously.
Tell your doctor if you notice any of the following and they worry you:
- insomnia
- disturbed sleep
- restlessness
- dizziness
- nausea
- headache
- anxiety
- agitation
- feeling of confusion
- diarrhoea
- vomiting
- paraesthesia
- constipation
- feeling of fullness
- upset stomach
- dry mouth
- blurred vision
- skin rash
- flushing
- hypotension
This is not a complete list of all possible side effects. Others may occur in some people and there may be some side effects not yet known.
Tell your doctor if you notice anything else that is making you feel unwell, even if it is not on this list.
Ask your doctor or pharmacist if you don’t understand anything in this list.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
After taking AURORIX
Storage
Keep AURORIX where young children cannot reach it.
A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Keep AURORIX in a cool dry place where the temperature stays below 30°C.
Do not store it, or any other medicine, in the bathroom or near a sink.
Do not leave it in the car or on window sills.
Heat and dampness can destroy some medicines.
Keep your tablets in the blister pack until it is time to take them.
If you take the tablets out of the blister, they may not keep well.
Disposal
If your doctor tells you to stop taking AURORIX, or the tablets have passed their expiry date, ask your pharmacist what to do with any tablets that are left over.
Product description
What AURORIX looks like
AURORIX comes in two strengths of tablets.
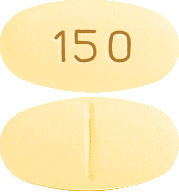
AURORIX 150 mg: oval, cylindrical, biconvex, pale yellow tablet marked "150" on one side with break bar on the other side.
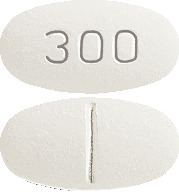
AURORIX 300 mg: oval, cylindrical, biconvex, white to yellow-white marked "300" on one side with break bar on the other side.
Ingredients
Each AURORIX 150 mg tablet contains 150 mg of the active ingredient moclobemide.
Each AURORIX 300 mg tablet contains 300 mg of the active ingredient moclobemide.
AURORIX 150 mg and 300 mg tablets also contain the following inactive ingredients:
- lactose monohydrate
- maize starch
- povidone
- sodium starch glycollate
- magnesium stearate
- ethylcellulose
- macrogol 6000
- hypromellose
- purified talc
- coloured with titanium dioxide.
AURORIX 150 mg tablets also contain yellow iron oxide.
AURORIX 150 mg: Contains sugars (as lactose).
AURORIX 300 mg: Contains lactose.
AURORIX tablets are gluten free.
AURORIX comes in boxes of 60 tablets.
Sponsor
Mylan Health Pty Ltd
Level 1, 30 The Bond
30-34 Hickson Road
Millers Point, NSW 2000
Australia
www.mylan.com.au
Phone: 1800 314 527
This leaflet was prepared in July 2021.
AURORIX 150 mg: AUST R 9987
AURORIX 300 mg: AUST R 51626
Version 8
Published by MIMS September 2021

 Other adverse events with an incidence of < 1% in clinical studies, or reported in postmarketing surveillance are as follows:
Other adverse events with an incidence of < 1% in clinical studies, or reported in postmarketing surveillance are as follows: The chemical name of moclobemide is p-chloro-N- (2-morpholinoethyl) benzamide. Its empirical formula is C13H17O2N2Cl with a molecular weight of 268.74.
The chemical name of moclobemide is p-chloro-N- (2-morpholinoethyl) benzamide. Its empirical formula is C13H17O2N2Cl with a molecular weight of 268.74.