1 Name of Medicine
Tacrolimus.
2 Qualitative and Quantitative Composition
Azematop 0.1% contains tacrolimus 1.0 mg/g (0.1%).
For full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
Azematop 0.1% is a white to slightly yellowish ointment.
4.1 Therapeutic Indications
Azematop 0.1% ointment is indicated for the treatment of moderate to severe atopic dermatitis in adults and adolescents (16 years of age and above).
Flare treatment.
Treatment of moderate to severe flares of atopic dermatitis in patients who are not adequately responsive to or are intolerant of conventional therapies such as topical corticosteroids.
Maintenance treatment.
Prevention of flares and prolongation of flare-free intervals in patients experiencing a high frequency of disease exacerbations who have responded to tacrolimus treatment during flares.4.2 Dose and Method of Administration
Azematop 0.1% treatment should be initiated by physicians with experience in the diagnosis and treatment of atopic dermatitis.
Dosage.
Azematop 0.1% can be used for short-term and intermittent long-term treatment. Treatment should not be continuous on a long-term basis.
Flare treatment.
Treatment should be started with Azematop 0.1% twice a day and treatment should be continued until clearance of the lesion. If symptoms recur, twice daily treatment with Azematop 0.1% should be restarted. An attempt should be made to reduce the frequency of application if the clinical condition allows.
Generally, improvement is seen within one week of starting treatment. If no signs of improvement are seen after two weeks of treatment, further treatment options should be considered.
Maintenance treatment.
Patients who are responding to up to 6 weeks treatment using tacrolimus ointment twice daily (lesions cleared, almost cleared or mildly affected) are suitable for maintenance treatment.
Azematop 0.1% ointment should be applied once a day twice weekly (e.g. Monday and Thursday) to areas commonly affected by atopic dermatitis to prevent progression to flares. Between applications there should be 2-3 days without Azematop 0.1% treatment.
After 12 months treatment, a review of the patient's condition should be conducted by the physician and a decision taken whether to continue maintenance treatment in the absence of safety data for maintenance treatment beyond 12 months.
If signs of a flare reoccur, twice daily treatment should be re-initiated (see Flare treatment section above).
Method of administration.
Azematop 0.1% ointment should be applied as a thin layer to affected or commonly affected areas of the skin. Azematop 0.1% ointment may be used on any part of the body, including face, neck and flexure areas, except on mucous membranes. Azematop 0.1% ointment should not be applied under occlusion because this method of administration has not been studied in patients (see Section 4.4).
Patients should be advised not to bathe, shower or swim immediately after applying the ointment; water may wash off the medicine.
Care should be taken to avoid contact with eyes and mucous membranes. If accidentally applied to these areas, the ointment should be thoroughly wiped off and/or rinsed off with water.
The use of tacrolimus ointment under occlusion has not been studied in patients. Occlusive dressings are not recommended.
As with any topical medicinal product, patients should wash their hands after application if the hands are not intended for treatment.4.3 Contraindications
Hypersensitivity to the active substance, macrolides in general, or to any of the excipients listed in Section 6.1.
4.4 Special Warnings and Precautions for Use
Exposure to sunlight/ultraviolet light.
Exposure of the skin to sunlight should be minimised and the use of ultraviolet (UV) light from a solarium, therapy with UVB or UVA in combination with psoralens (PUVA) should be avoided during use of Azematop 0.1% (see Section 5.3). Physicians should advise patients on appropriate sun protection methods, such as minimisation of the time in the sun, use of a sunscreen product and covering of the skin with appropriate clothing. Azematop 0.1% ointment should not be applied to lesions that are considered to be potentially malignant or pre-malignant.
The development of any new change different from previous eczema within a treated area should be reviewed by the physician.
Skin barrier defects and extensive disease.
The use of tacrolimus ointment is not recommended in patients with a skin barrier defect, such as Netherton's syndrome, lamellar ichthyosis, generalised erythroderma or cutaneous Graft Versus Host Disease. These skin conditions may increase systemic absorption of tacrolimus. Oral use of tacrolimus is also not recommended to treat these skin conditions. Post-marketing cases of increased tacrolimus blood level have been reported in these conditions. Azematop 0.1% should not be used in patients with congenital or acquired immunodeficiencies or in patients on therapy that cause immunosuppression.
Care should be exercised if applying Azematop 0.1% to patients with extensive skin involvement over an extended period of time.
Prolonged exposure.
Azematop 0.1% contains the active substance tacrolimus, a calcineurin inhibitor. In transplant patients, prolonged systemic exposure to intense immunosuppression following systemic administration of calcineurin inhibitors has been associated with an increased risk of developing lymphomas and skin malignancies.
Patients with atopic dermatitis treated with tacrolimus have not been found to have significant systemic tacrolimus levels and the role of local immunosuppression is unknown.
Based on the results of long-term studies and experience a link between Azematop 0.1% ointment treatment and development of malignancies has not been confirmed, but definitive conclusions cannot be drawn. It is recommended to use tacrolimus ointment at the lowest strength and the lowest frequency for the shortest duration necessary as determined by the physician's evaluation of the clinical condition (see Section 4.2).
Lymphadenopathy.
Lymphadenopathy was uncommonly (0.8%) reported in clinical trials. The majority of these cases were related to infections (skin, respiratory tract, tooth) and resolved with appropriate antibiotic therapy.
Lymphadenopathy present at initiation of therapy should be investigated and kept under review. In case of persistent lymphadenopathy, the aetiology of the lymphadenopathy should be investigated. In the absence of a clear aetiology for the lymphadenopathy or in the presence of acute infectious mononucleosis, discontinuation of Azematop 0.1% should be considered. Patients who develop lymphadenopathy during treatment should be monitored to ensure that the lymphadenopathy resolves.
Superficial skin infections.
Patients with atopic dermatitis are predisposed to superficial skin infections.
Tacrolimus ointment has not been evaluated for its efficacy and safety in the treatment of clinically infected atopic dermatitis. Before commencing treatment with Azematop 0.1% ointment, clinical infections at treatment sites should be cleared. Treatment with Azematop 0.1% may be associated with an increased risk of folliculitis and herpes viral infections (herpes simplex dermatitis [eczema herpeticum], herpes simplex [cold sores], Kaposi's varicelliform eruption) (see Section 4.8). In the presence of these infections, the balance of risks and benefits associated with Azematop 0.1% use should be evaluated.
Concomitant use of other topical preparations.
Formal topical drug interaction studies with tacrolimus ointment have not been conducted.
Emollients should not be applied to the same area within 2 hours of applying Azematop 0.1% ointment. Concomitant use of other topical preparations has not been assessed. There is no experience with concomitant use of systemic steroids or immunosuppressive agents.
Use in hepatic impairment.
Tacrolimus is extensively metabolised in the liver and although blood concentrations are low following topical therapy, the ointment should be used with caution in patients with hepatic failure (see Section 5.2).
Use in the elderly.
Specific studies have not been conducted in older people. However, the clinical experience available in this patient population has not shown the necessity for any dosage adjustment.
Paediatric use.
Azematop 0.1% ointment should not be used in children aged below 16 years until further data are available.
Effects on laboratory tests.
No data are available.4.5 Interactions with Other Medicines and Other Forms of Interactions
Tacrolimus is not metabolised in human skin, indicating that there is no potential for percutaneous interactions that could affect the metabolism of tacrolimus.
Systemically available tacrolimus is metabolised via the hepatic cytochrome P450 3A4 (CYP3A4). Systemic exposure from topical application of tacrolimus ointment is low (< 1.0 nanogram/mL) and is unlikely to be affected by concomitant use of substances known to be inhibitors of CYP3A4. However, the possibility of interactions cannot be ruled out and the concomitant systemic administration of known CYP3A4 inhibitors (e.g. erythromycin, itraconazole, ketoconazole and diltiazem) in patients with widespread and/or erythrodermic disease should be done with caution.
4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
Tacrolimus subcutaneously administered to male rats at doses of 2 or 3 mg/kg/day resulted in a dose-related decrease in sperm count.
(Category C)
In reproduction studies in rats and rabbits, adverse effects on the foetus were observed mainly at dose levels that were toxic to the dams.
Tacrolimus given orally at 1.0 mg/kg (0.8 to 2.2 times the clinical dose range based on body surface area) to male and female rats, prior to and during mating, as well as to dams during gestation and lactation, was associated with embryolethality and adverse effects on female reproduction which were indicated by a higher rate of post-implantation loss and increased numbers of undelivered and nonviable pups. When given at 3.2 mg/kg (2.6 to 6.9 times the clinical dose range based on body surface area), tacrolimus was associated with maternal and paternal toxicity as well as reproductive toxicity including marked adverse effects on estrus cycles, parturition, pup viability, and pup malformations.
Human data show that tacrolimus is able to cross the placenta.
There are no adequate and well-controlled studies in pregnant women. There is data from a voluntary pregnancy exposure registry (Transplantation Pregnancy Registry International (TPRI)) that monitors outcomes of pregnancy in female transplant recipients and those fathered by male transplant recipients exposed to immunosuppressants including tacrolimus.
Safety data from post-marketing surveillance and the TPRI suggest infants of organ transplant recipients using immunosuppressive medications including oral tacrolimus have an increased risk for miscarriage, pre-term delivery (< 37 weeks), low birth weight (< 2500 g), birth defects/congenital anomalies and foetal distress.
The use of oral tacrolimus for systemic immunosuppression during pregnancy has been associated with neonatal hyperkalaemia and renal dysfunction.
Systemic tacrolimus use may also be associated with hyperglycaemia or diabetes in pregnancy in the absence of pre-existing diabetes or gestational diabetes.
Systemic tacrolimus may exacerbate hypertension in pregnant women and increase pre-eclampsia.
Females and males of reproductive potential should consider the use of appropriate contraception prior to starting treatment with tacrolimus.
Tacrolimus should be used during pregnancy only if the potential benefit to the mother justifies potential risk to the foetus.
Tacrolimus is excreted into breast milk. It is therefore recommended that mothers should not breast-feed while receiving tacrolimus.4.7 Effects on Ability to Drive and Use Machines
Azematop 0.1% ointment has no or negligible influence on the ability to drive or use machines.
4.8 Adverse Effects (Undesirable Effects)
Adverse events.
Table 1 depicts the adjusted incidence of adverse events pooled across the 3 identically designed 12-week controlled studies for patients in vehicle, tacrolimus ointment 0.03%, and tacrolimus ointment 0.1% treatment groups. The table also depicts the unadjusted incidence of adverse events in four safety studies, regardless of relationship to study drug.
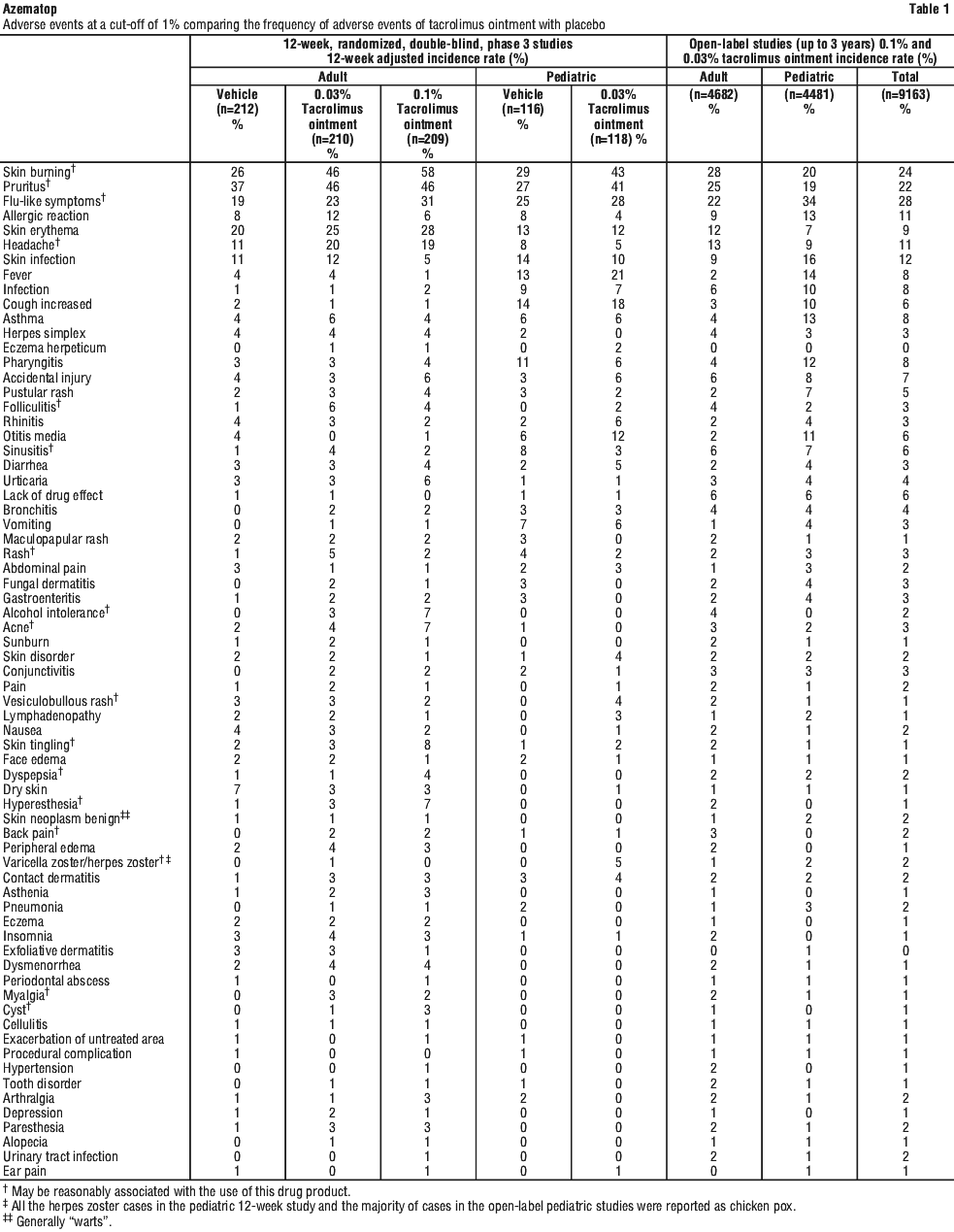 Other adverse events which occurred at an incidence between 0.2% and less than 1% in clinical studies in the above table include: abnormal vision, abscess, anaphylactoid reaction, anemia, anorexia, anxiety, arthritis, arthrosis, bilirubinemia, blepharitis, bone disorder, breast neoplasm benign, bursitis, cataract NOS, chest pain, chills, colitis, conjunctival edema, constipation, cramps, cutaneous moniliasis, cystitis, dehydration, dizziness, dry eyes, dry mouth/nose, dyspnea, ear disorder, ecchymosis, edema, epistaxis, eye pain, furunculosis, gastritis, gastrointestinal disorder, hernia, hypercholesterolemia, hypertonia, hypothyroidism, joint disorder, laryngitis, leukoderma, lung disorder, malaise, migraine, moniliasis, mouth ulceration, nail disorder, neck pain, neoplasm benign, oral moniliasis, otitis externa, photosensitivity reaction, rectal disorder, seborrhea, skin carcinoma, skin discoloration, skin hypertrophy, skin ulcer, stomatitis, tendon disorder, thinking abnormal, tooth caries, sweating, syncope, tachycardia, taste perversion, unintended pregnancy, vaginal moniliasis, vaginitis, valvular heart disease, vasodilatation, and vertigo.
Other adverse events which occurred at an incidence between 0.2% and less than 1% in clinical studies in the above table include: abnormal vision, abscess, anaphylactoid reaction, anemia, anorexia, anxiety, arthritis, arthrosis, bilirubinemia, blepharitis, bone disorder, breast neoplasm benign, bursitis, cataract NOS, chest pain, chills, colitis, conjunctival edema, constipation, cramps, cutaneous moniliasis, cystitis, dehydration, dizziness, dry eyes, dry mouth/nose, dyspnea, ear disorder, ecchymosis, edema, epistaxis, eye pain, furunculosis, gastritis, gastrointestinal disorder, hernia, hypercholesterolemia, hypertonia, hypothyroidism, joint disorder, laryngitis, leukoderma, lung disorder, malaise, migraine, moniliasis, mouth ulceration, nail disorder, neck pain, neoplasm benign, oral moniliasis, otitis externa, photosensitivity reaction, rectal disorder, seborrhea, skin carcinoma, skin discoloration, skin hypertrophy, skin ulcer, stomatitis, tendon disorder, thinking abnormal, tooth caries, sweating, syncope, tachycardia, taste perversion, unintended pregnancy, vaginal moniliasis, vaginitis, valvular heart disease, vasodilatation, and vertigo.
Maintenance treatment adverse events.
In a study of maintenance treatment (twice weekly treatment) in adults and children with moderate and severe atopic dermatitis the following adverse events were noted to occur more frequently than in the control group: application site impetigo (7.7% in children) and application site infections (6.4% in children and 6.3% in adults).
In the two Phase 3, multi-centre, double-blind, vehicle-controlled 12-month studies the nature and incidence of adverse events were consistent with the established safety profile of tacrolimus ointment.
Table 2 describes the most frequently reported adverse events (≥ 3%) that occurred in the Phase 3 study in adults.
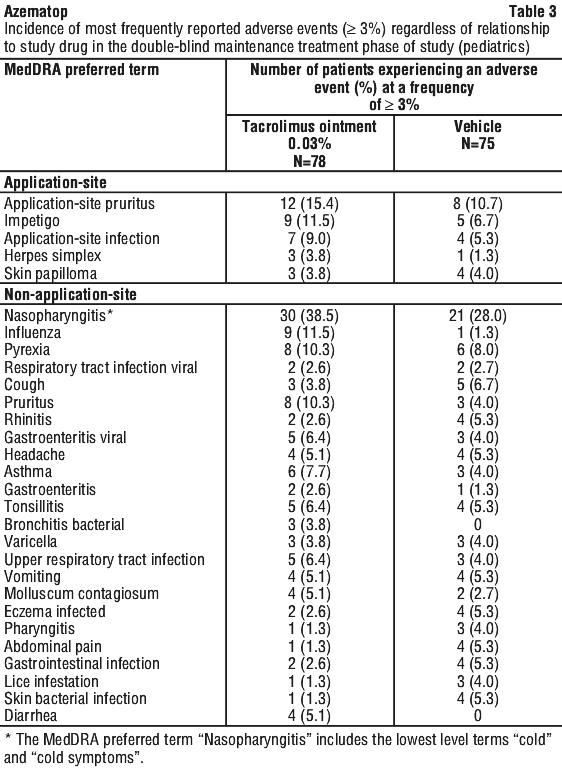
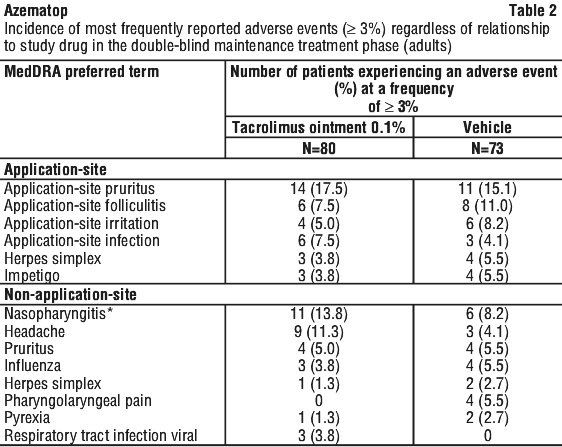 Table 3 describes the most frequently reported adverse events (≥ 3%) that occurred in the Phase 3 study in pediatrics.
Table 3 describes the most frequently reported adverse events (≥ 3%) that occurred in the Phase 3 study in pediatrics.
Adverse reactions.
In clinical studies approximately 50% of patients experienced some type of skin irritation adverse reaction at the site of application. Burning sensation and pruritus were very common, usually mild to moderate in severity and tended to resolve within one week of starting treatment. Erythema was a common skin irritation adverse reaction. Sensation of warmth, pain, paraesthesia and rash at the site of application were also commonly observed. Alcohol intolerance (facial flushing or skin irritation after consumption of an alcoholic beverage) was common. Patients may be at an increased risk of folliculitis, acne and herpes viral infections.
Adverse reactions with suspected relationship to treatment are listed below by system organ class. Frequencies are defined as very common (≥ 1/10), common (≥ 1/100 to < 1/10) and uncommon (≥ 1/1,000 to < 1/100). Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness.
Infections and infestations.
Common: local skin infection regardless of specific aetiology including but not limited to: eczema herpeticum, folliculitis, herpes simplex, herpes virus infection.
Metabolism and nutrition disorders.
Common: alcohol intolerance (facial flushing or skin irritation after consumption of an alcoholic beverage).
Nervous system disorders.
Common: paraesthesias and dysaesthesias (hyperaesthesia, burning sensation).
Skin and subcutaneous tissue disorders.
Common: pruritus.
General disorders and administration site conditions.
Very common: application site burning, application site pruritus.
Common: application site warmth, application site erythema, application site pain, application site irritation, application site paraesthesia, application site rash.
Post-marketing adverse reactions.
Cases of malignancies, including cutaneous (i.e. cutaneous T cell lymphomas) and other types of lymphoma, and skin cancers, have been reported in patients using tacrolimus ointment (see Section 4.4).
The following adverse reactions have been identified during post-approval use of tacrolimus ointment. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
CNS.
Seizures.
Infections and infestations.
Common: Kaposi's varicelliform eruption.
Not known: ophthalmic herpes infection.
Bullous impetigo, osteomyelitis, septicemia, skin infection regardless of specific etiology.
Neoplasms.
Lymphomas, basal cell carcinoma, squamous cell carcinoma, malignant melanoma.
Renal.
Acute renal failure in patients with or without Netherton's syndrome, renal impairment.
Skin.
Un-common: acne.
Not known: rosacea, lentigo, application site edema.
Investigations.
Not known: drug level increased.
Reporting suspected adverse effects.
Information on how to report adverse events:
reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at www.tga.gov.au/reporting-problems.4.9 Overdose
Overdosage following topical administration is unlikely.
If ingested, general supportive measures may be appropriate. These may include monitoring of vital signs and observation of clinical status. Due to the nature of the ointment vehicle, induction of vomiting or gastric lavage is not recommended.
5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: other dermatological preparations, ATC code: D11AH01.
Mechanism of action.
The mechanism of action of tacrolimus in atopic dermatitis is not fully understood. While the following have been observed, the clinical significance of these observations in atopic dermatitis is not known.
Via its binding to a specific cytoplasmic immunophilin (FKBP12), tacrolimus inhibits calcium-dependent signal transduction pathways in T cells, thereby preventing the transcription and synthesis of IL-2, IL-3, IL-4, IL-5 and other cytokines such as GM-CSF, TNF-α and IFN-γ. In vitro, in Langerhans cells isolated from normal human skin, tacrolimus reduced the stimulatory activity towards T cells. Tacrolimus has also been shown to inhibit the release of inflammatory mediators from skin mast cells, basophils and eosinophils.
In animals, tacrolimus ointment suppressed inflammatory reactions in experimental and spontaneous dermatitis models that resemble human atopic dermatitis. Tacrolimus ointment did not reduce skin thickness and did not cause skin atrophy in animals.
In patients with atopic dermatitis, improvement of skin lesions during treatment with tacrolimus ointment was associated with reduced Fc receptor expression on Langerhans cells and a reduction of their hyperstimulatory activity towards T cells. Tacrolimus ointment does not affect collagen synthesis in humans.
Clinical trials.
The efficacy and safety of tacrolimus ointment was assessed with the innovator tacrolimus ointments in Phase I to Phase III clinical trials. Data from major trials are presented here.
A prospective, randomised, investigator blinded, multicentre comparative trial compared the efficacy of 0.1% tacrolimus ointment (Protopic; Astellas Pharma US Inc.) and 1% pimecrolimus cream (Elidel; Novartis Pharmaceuticals Corp.) in a total of adult 281 patients (141 treated with tacrolimus and 140 treated with pimecrolimus) with moderate to severe atopic dermatitis. Patient's ≥ 16 years of age were eligible for enrolment if they met the Hanifin and Rajka clinical criteria for the diagnosis of atopic dermatitis (AD). Patients were examined at baseline/day 1, week 1, week 3, and week 6/EOS for efficacy and safety. The primary end point was the percent change in the EASI score from baseline to week 6/EOS. By week 3 and continuing to the study end, there were significantly greater improvements in the EASI score in the tacrolimus treatment group compared with the pimecrolimus treatment group (a reduction of 57% vs 39% at study end). Likewise, success with therapy (defined as an IGADA of clear or almost clear) occurred in significantly more tacrolimus treated patients than pimecrolimus-treated patients (40% vs 22% at study end). With regard to improvement in %BSA affected, there was also a significantly greater improvement in the tacrolimus treatment group compared with the pimecrolimus treatment group beginning with week 3.
A phase III comparative, multicentre, randomised, double-blind, parallel-group study was undertaken to compare 0.03% and 0.1% tacrolimus (Protopic) ointment with 0.1% hydrocortisone-17-butyrate ointment, a mid-potent to potent topical corticosteroid, in the treatment of adult patients with moderate-to severe atopic dermatitis. Male and female patients, 16 to 70 years old, with a diagnosis of AD on the basis of the criteria of Hanifin and Rajka were eligible for study participation. The mEASI mAUC as a percentage of baseline showed that, averaged over the 3-week course of treatment, patients had a median improvement of 53.0% with 0.03% tacrolimus, 63.5% with 0.1% tacrolimus, and 63.9% with hydrocortisone butyrate
In a six-month multicentre double-blind randomised trial, 0.1% tacrolimus ointment was administered twice-a-day to adults with moderate to severe atopic dermatitis and compared to a topical corticosteroid-based regimen (0.1% hydrocortisone butyrate on trunk and extremities, 1% hydrocortisone acetate on face and neck). The primary endpoint was the response rate at month 3 defined as the proportion of patients with at least 60% improvement in the mEASI (modified Eczema Area and Severity Index) between baseline and month 3. By month 3, more patients in the 0.1% tacrolimus group responded to treatment (72.6% vs. 52.3% in the corticosteroid group, P < 0.001). The patients treated with 0.1% tacrolimus also showed greater improvement in mEASI, EASI, affected body surface area and physician and patient assessments of global response.
An international, multicentre, randomised, double-blind phase IV trial was conducted at 108 centres in Belgium, Finland, France, Morocco, Romania and Tunisia to compare the efficacy of tacrolimus 0.1% and fluticasone 0.005% ointments on facial lesions. Patients aged ≥ 16 years with moderate to severe AD were randomised to treatment of facial lesions with twice-daily tacrolimus 0.1% ointment or fluticasone 0.005% ointment for 3 weeks or until clearance. The primary endpoint was the response rate, defined as the percentage of patients with at least 60% reduction in the modified Local Eczema and Severity Index (mLEASI) score from baseline to day 21. Response rate in the FAS was 93.3% with tacrolimus ointment and 87.8% with fluticasone. Substantial reductions in severity at day 21 were observed for individual signs of AD in both treatment groups. The physicians' global assessment of clinical response for the facial region was significantly different between the two groups; for example, 250/283 patients (88%) in the tacrolimus ointment group and 220/279 (79%) in the fluticasone ointment group showed marked or excellent improvement, or clearance of lesions. In conclusion, tacrolimus 0.1% ointment has superior efficacy to fluticasone 0.005% ointment for twice-daily treatment of adults with moderate to severe facial AD in whom conventional therapy was inadequately effective or not tolerated.
The efficacy and safety of tacrolimus ointment in maintenance treatment of mild to severe atopic dermatitis was assessed in a Phase III multicentre clinical trial in adult patients (≥ 16 years). In the study, patients with active disease entered an open-label period (OLP) during which they treated affected lesions with tacrolimus ointment twice daily until improvement had reached a predefined score (Investigator's Global Assessment [IGA] ≤ 2, i.e. clear, almost clear or mild disease) for a maximum of 6 weeks. Thereafter, patients entered a double-blind disease control period (DCP) for up to 12 months. Patients were randomised to receive either tacrolimus ointment (0.1%) or vehicle, once a day twice weekly. If a disease exacerbation occurred, patients were treated with open-label tacrolimus ointment twice daily for a maximum of 6 weeks until the IGA score returned to ≤ 2.
The primary endpoint was the number of major flares during the DCP (major flares were defined as disease exacerbations with an IGA > 2 on day 1 of the exacerbation that required the use of twice-daily tacrolimus ointment for > 7 days). A significant benefit with twice weekly treatment with tacrolimus ointment was reported with regard to the primary and key secondary endpoints over a period of 12 months in a pooled population of patients with mild to severe atopic dermatitis.
5.2 Pharmacokinetic Properties
Clinical data have shown that tacrolimus concentrations in systemic circulation after topical administration are low and, when measurable, transient.
Absorption.
Data from healthy human subjects indicate that there is little or no systemic exposure to tacrolimus following single or repeated topical application of tacrolimus ointment.
Target trough concentrations for systemic immunosuppression for oral tacrolimus are 5-20 nanogram/mL in transplant patients. Most adults with atopic dermatitis treated with single or repeated application of tacrolimus ointment (0.1%) had blood concentrations < 1.0 nanogram/mL. When observed, blood concentrations exceeding 1.0 nanogram/mL were transient. Systemic exposure increases with increasing treatment areas. However, both the extent and the rate of topical absorption of tacrolimus decrease as the skin heals. In adults with an average of 50% body surface area treated, systemic exposure (i.e. AUC) of tacrolimus from tacrolimus ointment is approximately 30-fold less than that seen with oral immunosuppressive doses in kidney and liver transplant patients. The lowest tacrolimus blood concentration at which systemic effects can be observed is not known.
There was no evidence of systemic accumulation of tacrolimus in adults treated for prolonged periods (up to one year) with tacrolimus ointment.
Distribution.
In the systemic circulation, tacrolimus binds strongly to erythrocytes resulting in an approximate 20:1 distribution ratio of whole blood/plasma concentrations. In plasma, tacrolimus is highly bound (> 98.8%) to plasma proteins, mainly to serum albumin and α-1-acid glycoprotein.
Metabolism.
Systemically available tacrolimus is extensively metabolised in the liver via CYP3A4.
Excretion.
When administered intravenously, tacrolimus has been shown to have a low clearance rate. The average total body clearance is approximately 2.25 L/h. The hepatic clearance of systemically available tacrolimus could be reduced in subjects with severe hepatic impairment, or in subjects who are co-treated with drugs that are potent inhibitors of CYP3A4.
Following repeated topical application of the ointment the average half-life of tacrolimus was estimated to be 75 hours for adults and 65 hours for children.
Paediatric population.
The pharmacokinetics of tacrolimus after topical application are similar to those reported in adults, with minimal systemic exposure and no evidence of accumulation (see above).
5.3 Preclinical Safety Data
Genotoxicity.
No evidence of genotoxicity was seen in a series of assays for gene mutations and clastogenicity. Tacrolimus did not cause unscheduled DNA synthesis in rodent hepatocytes but high concentrations of tacrolimus have been reported to increase the frequency of sister chromatid exchanges in human lymphocytes in vitro.
Carcinogenicity.
Oral (feed) carcinogenicity studies have been carried out with systemically administered tacrolimus in male and female rats and mice. In the 80-week mouse study and in the 104-week rat study no relationship of tumour incidence to tacrolimus dosage was found at daily doses up to 3 mg/kg (9 x the maximum recommended human dose (MRHD) based on AUC comparisons) and 5 mg/kg (3 x the MRHD based on AUC comparisons), respectively.
A 104-week dermal carcinogenicity study was performed in mice with tacrolimus ointment (0.03-3%, equivalent to tacrolimus doses of 1.1-1.18 mg/kg/day or 3.3-354 mg/m2/day. In the study, the incidence of skin tumours was minimal, and the topical application of tacrolimus was not associated with skin tumour formation under ambient room lighting. However, a statistically significant elevation in the incidence of pleomorphic lymphoma was reported in high dose male (25/50) and female animals (27/50) and in the incidence of undifferentiated lymphoma in high dose female animals (13/50) was noted in the mouse dermal carcinogenicity study. Lymphomas were noted in the mouse dermal carcinogenicity study at a daily dose of 3.5 mg/kg (0.1% tacrolimus ointment) (26 x MRHD based on AUC comparisons). No drug-related tumours were noted in the mouse dermal carcinogenicity study at a daily dose of 1.1 mg/kg (0.03% tacrolimus ointment) (10 x MRHD based on AUC comparisons).
In a 52-week photocarcinogenicity study, the median time to onset of skin tumour formation was decreased in hairless mice following chronic topical dosing with concurrent exposure to UV radiation (40 weeks of treatment followed by 12 weeks of observation) with tacrolimus ointment at ≥ 0.1% tacrolimus.6 Pharmaceutical Particulars
6.1 List of Excipients
Liquid paraffin, white soft paraffin, propylene carbonate, white beeswax, hard paraffin.
6.2 Incompatibilities
Not applicable.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiry date can be found on the packaging.
The product should be used within 3 months after opening.
6.4 Special Precautions for Storage
Store below 25°C.
6.5 Nature and Contents of Container
Aluminium laminate tube with low-density-polyethylene inner coat fitted with a white polypropylene screw cap.
Package sizes registered: 10 g, 30 g and 60 g. Some registered pack sizes may not be sold in Australia.
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties

CAS number.
104987-11-3.7 Medicine Schedule (Poisons Standard)
Prescription only medicine (S4).
Summary Table of Changes
