What is in this leaflet
Read this leaflet carefully before taking Baraclude. This leaflet answers some common questions about Baraclude.
It does not contain all the available information.
It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you taking Baraclude against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may need to read it again.
What Baraclude is used for
Baraclude contains entecavir and belongs to a group of medicines called antiviral medicines.
Baraclude is used to treat adults infected with hepatitis B virus.
How Baraclude Works
Infection by the hepatitis B virus can lead to damage to the liver. Baraclude reduces the amount of virus in your body, and has been shown to improve the condition of the liver.
It is not known how safe Baraclude is when taken for long periods.
Your doctor may have prescribed Baraclude for another reason. Ask your doctor if you have any questions about why Baraclude has been prescribed for you.
Baraclude is not addictive. This medicine is available only with a doctor's prescription.
Baraclude is not recommended for use in children under 16 years, as there have been no studies of its effects in children.
Before you take Baraclude
It is important that you check the information below before you take Baraclude.
When you must not take Baraclude
You must not take Baraclude if you have a history of severe allergic reactions to Baraclude or to any of the ingredients listed at the end of this leaflet. Symptoms of a severe allergic reaction may include; chills, fever, fast heart beat, wheezing or coughing, difficulty breathing, dizziness, flushing, sweating and swelling of the face, tongue or other parts of the body.
Do not use Baraclude after the expiry date printed on the back of the pack. If this medicine is taken after the expiry date has passed, it may not work as well.
Do not take Baraclude if the packaging is torn or shows signs of tampering.
Before you start to take Baraclude
It is important to remain under the care of your doctor during Baraclude therapy and after stopping Baraclude. You should report any new symptoms, medications or any other aspects affecting your health to your doctor. Your hepatitis B virus infection may get worse if you stop taking Baraclude. If your doctor advises you to stop Baraclude, they will monitor your health and perform regular blood tests to monitor your liver.
Tell your doctor if you:
- have allergies to:
- any other medicines you have been given or purchased
- substances such as foods, preservatives or dyes;
Symptoms of a severe allergic reaction may include; chills, fever, fast heart beat, wheezing or coughing, difficulty breathing, dizziness, flushing, sweating and swelling of the face, tongue or other parts of the body.
- are pregnant or intend to become pregnant.
Experience is limited with the use of Baraclude in pregnant women. Therefore, it should not be used during pregnancy unless it is clearly needed. If there is an urgent need to consider Baraclude during pregnancy, your doctor will discuss with you the benefits and risks of taking it. If you take Baraclude while you are pregnant, talk to your doctor about how you can take part in the Baraclude Pregnancy Registry. The purpose of the pregnancy registry is to collect information about the health of you and your baby.
- are breast feeding or planning to breast-feed.
It is not known whether Baraclude passes into breast milk. Therefore to avoid possible side effects in the nursing infant, mothers should stop breast-feeding if they are taking Baraclude;
- currently experience or have experienced any medical conditions especially any problems with your kidneys.
- have HIV and you are not currently on HIV treatment.
Baraclude is not recommended in patients who have both HIV and Hepatitis B and who are not currently receiving anti-HIV treatment. Baraclude may affect your HIV virus which could impact on future treatment options for HIV.
- are lactose intolerant. Baraclude tablets contain lactose. Baraclude tablets should be used with caution in patients who are lactose intolerant. Speak to your doctor if you are lactose intolerant.
If you have not told your doctor about any of the above, tell them before you use Baraclude.
Taking other medicines
Tell your doctor if you are taking other medicines, including vitamin supplements, herbal preparations or any medicines you buy with or without a prescription from your pharmacy, supermarket or health food shop.
Your doctor and pharmacist may have more information on medicines to be careful with, or to avoid while taking Baraclude.
How to take Baraclude
Baraclude should be given only when prescribed by your doctor. Follow all directions given to you by your doctor or pharmacist carefully. They may differ from the information contained in this leaflet.
How much to take
The usual dose of Baraclude is 0.5 mg (one white tablet) or 1 mg (one pink tablet) once a day.
If you have a medical problem with your kidneys your doctor may need to change how often you take your Baraclude tablets.
Your doctor will tell you what dose to take and how often you should take your Baraclude tablets.
Please talk to your doctor or pharmacist for more information.
How to take it
Swallow the tablet whole with a glass of water. The dose of Baraclude should be taken on an empty stomach.
When to take Baraclude
Baraclude may be taken at any time of day provided it is taken on an empty stomach. Empty stomach means at least 2 hours after a meal and at least 2 hours before the next meal.
Talk to your doctor or pharmacist to work out when it is best for you to take your dose of Baraclude.
How long to take it
Baraclude helps control your condition but does not cure it. Therefore you must take Baraclude every day as directed by your doctor. Continue taking Baraclude for as long as your doctor tells you to.
Your doctor has prescribed Baraclude to prevent hepatitis B virus from further damaging your liver.
Baraclude is a very important treatment that can improve the inflammation and scar tissue caused by the hepatitis B virus in your liver and may reduce the chance of developing cirrhosis, liver failure and liver cancer.
It is extremely important that you do not stop taking Baraclude without discussing it with your doctor. If Baraclude is suddenly stopped, the hepatitis B virus can become very active again and lead to sudden development of severe liver failure. There is a high risk of dying if liver failure develops and liver transplantation may be necessary to save your life.
It is important to take Baraclude every day or as directed by your doctor, to not miss medicine doses, and to make sure you have enough supply until you next see your doctor.
Do not stop taking Baraclude or change the dose unless asked to do so by your doctor, even if you feel better, as it can be very dangerous.
If you forget to take it
If it is almost time for your next dose, skip the dose you missed and take the next dose when you are meant to.
Otherwise take it as soon as you remember, and then go back to taking it as you would normally.
Do not take a double dose to make up for the dose you missed. This may increase the chance of you getting an unwanted side effect. If you are not sure what to do, ask your doctor or pharmacist.
If you have trouble remembering when to take your medicine, ask your pharmacist for some hints and inform your doctor that you have missed a dose. It is very important not to miss your doses of Baraclude.
If you take too much (overdose)
Immediately call your doctor or the Poisons Information Centre on 131126 in Australia or 0800 764 766 in New Zealand, or go to the Accident and Emergency Centre at your nearest hospital if you or anyone else takes too much Baraclude. Do this even if there are no signs of discomfort or poisoning.
While you are using Baraclude
Things you must do
- If you become pregnant while taking Baraclude, tell your doctor immediately.
- If you are about to start taking any new medicines, tell your doctor and pharmacist that you are taking Baraclude. Baraclude may interfere with the medicine you are taking.
- If you are about to have any medical tests, tell your doctor that you are taking Baraclude. Baraclude may interfere with the results of these tests.
- If you plan to have surgery, tell your doctor or dentist that you are taking Baraclude.
Things you must not do
- Do not give Baraclude to anyone else, even if they have the same condition as you.
- Do not use Baraclude to treat any other complaints unless your doctor tells you to.
- Do not stop taking Baraclude or lower the dosage without checking with your doctor. Your hepatitis may worsen after stopping treatment.
Things to be careful of
- Be careful driving or operating machinery until you know how Baraclude affects you.
Some patients taking Baraclude have experienced dizziness. It is not known if this was caused by Baraclude. Make sure you know how you react to Baraclude before you drive a car, operate machinery or do anything else that could be dangerous if you are dizzy. - Make sure that you visit your doctor regularly throughout your entire course of treatment with Baraclude.
When your treatment with Baraclude is stopped, your doctor will continue to monitor you and take blood tests for several months. - There is no evidence that Baraclude reduces the risk of infecting others with hepatitis B through sexual contact or body fluids (including blood contamination).
Therefore it is important to take appropriate precautions to prevent others being infected with hepatitis B. Talk to your doctor about safe sexual practices that protect your partner. Never share needles. Do not share personal items that can have blood or bodily fluids on them, like toothbrushes and razor blades. A vaccine is available to protect those at risk of becoming infected with hepatitis B.
Side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Baraclude. Baraclude helps most people with hepatitis B infection but it may have unwanted side effects in some people. Ask your doctor or pharmacist to answer any questions you may have.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. Some very important side effects are listed below.
Tell your doctor immediately, or go to accident and emergency at your nearest hospital if you notice any of the following signs of a sudden life-threatening allergic reaction:
chills, fever, fast heart beat, wheezing or coughing, difficulty breathing, dizziness, flushing, sweating and swelling of the face, tongue or other parts of the body.
Some people who have taken Baraclude or medicines like Baraclude have developed a serious condition called lactic acidosis. Lactic acidosis is a serious medical emergency that can cause death. Lactic acidosis must be treated in the hospital. Reports of lactic acidosis with Baraclude generally involved patients who were seriously ill due to their liver disease or other medical condition.
Call your healthcare provider right away if you get any of the following signs or symptoms of lactic acidosis:
Feeling very weak or tired, unusual muscle pain, trouble breathing, stomach pain with nausea and vomiting, feeling cold (especially in your arms and legs), feeling dizzy or light-headed, fast or irregular heartbeat.
Some people who have taken medicines like Baraclude have developed serious liver problems called hepatotoxicity, with liver enlargement (hepatomegaly) and fat in the liver (steatosis). Hepatomegaly with steatosis is a serious medical emergency that can cause death.
Call your healthcare provider right away if you get any of the following signs or symptoms of liver problems:
Your skin or the white part of your eyes turns yellow (jaundice), urine turns dark, bowel movements (stools) turn light in colour, you don't feel like eating food for several days or longer, nausea, lower stomach pain. You may be more likely to get lactic acidosis or serious liver problems if you are female, very overweight, or have been taking medicines, like Baraclude, for a long time.
The most common side-effects are diarrhoea, indigestion, tiredness and headache.
This is not a complete list of side effects, other side effects not listed above may also occur in some patients.
Tell your doctor if you notice anything that is making you feel unwell.
Do not be alarmed by this list of possible side effects. You may not experience any of them or only some of them.
After using Baraclude
Storage
Store Baraclude tablets in a cool dry place where the temperature stays below 30°C.
Keep your tablets in the blister pack until it is time to take one. If you take the tablets out of the pack they may not keep as well.
Do not store Baraclude or any other medicine in the bathroom or near the kitchen sink. Do not leave it in the car. Heat and dampness can destroy some medicines.
Do not keep Baraclude tablets where children can reach them. A locked cupboard at least one-and-a-half metres above the ground is a good place to store medicines.
Disposal
If your doctor tells you to stop taking Baraclude, or the tablets have passed their expiry date, ask your pharmacist what to do with what is left over.
Product description
What it looks like
Baraclude tablets come in two types:
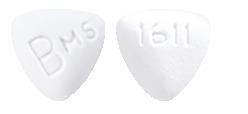
- Baraclude 0.5 mg tablet - white, triangular shaped tablets with 'BMS' on one side and '1611' on the other
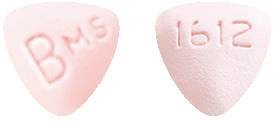
- Baraclude 1 mg tablet - pink triangular shaped tablets with 'BMS' on one side and '1612' on the other
Ingredients
Each tablet contains:
Active ingredients:
- Baraclude 0.5 mg tablet - 0.5 mg of entecavir per tablet
- Baraclude 1 mg tablet - 1 mg of entecavir per tablet
Other ingredients:
lactose monohydrate, microcrystalline cellulose, crospovidone, povidone, and magnesium stearate. The 0.5 mg tablet coating contains titanium dioxide, hypromellose, Macrogol 400, polysorbate 80, and the 1 mg tablet coating contains titanium dioxide, hypromellose, Macrogol 400 and iron oxide red CI177491.
Registration Numbers
Baraclude 0.5 mg - 30's - AUST R 116852
Baraclude 1 mg - 30's - AUST R 116853
Sponsored by
Bristol-Myers Squibb Australia Pty Ltd,
4 Nexus Court, Mulgrave,
Victoria 3170, Australia
Date of Preparation: March 2020
This information in no way replaces the advice of your doctor or pharmacist.
Baraclude is a Trademark of a Bristol-Myers Squibb Company.
Published by MIMS May 2020


 Among Baraclude-treated patients in these studies, on-treatment ALT elevations > 10 X ULN and > 2 X baseline generally resolved with continued treatment. A majority of these exacerbations were associated with a > 2 log10/mL reduction in viral load that preceded or coincided with the ALT elevation. Periodic monitoring of hepatic function is recommended during treatment.
Among Baraclude-treated patients in these studies, on-treatment ALT elevations > 10 X ULN and > 2 X baseline generally resolved with continued treatment. A majority of these exacerbations were associated with a > 2 log10/mL reduction in viral load that preceded or coincided with the ALT elevation. Periodic monitoring of hepatic function is recommended during treatment.
 Causes of death in Study AI463048 were generally liver related, as expected in this population. The time to onset of HCC or death (whichever occurred first) was comparable in the two treatment groups.
Causes of death in Study AI463048 were generally liver related, as expected in this population. The time to onset of HCC or death (whichever occurred first) was comparable in the two treatment groups.



 Responses for patients with baseline Knodell Fibrosis Scores of 4 (cirrhosis) were comparable to overall responses on all efficacy outcome measures (all patients had compensated liver disease). Histologic improvement was independent of baseline HBV DNA or ALT levels.
Responses for patients with baseline Knodell Fibrosis Scores of 4 (cirrhosis) were comparable to overall responses on all efficacy outcome measures (all patients had compensated liver disease). Histologic improvement was independent of baseline HBV DNA or ALT levels.
 In Study AI463026, responses for patients with baseline Knodell Fibrosis Scores of 4 (cirrhosis) were comparable to overall responses on all efficacy outcome measures (all patients had compensated liver disease). Histologic Improvement was independent of baseline HBV DNA or ALT levels.
In Study AI463026, responses for patients with baseline Knodell Fibrosis Scores of 4 (cirrhosis) were comparable to overall responses on all efficacy outcome measures (all patients had compensated liver disease). Histologic Improvement was independent of baseline HBV DNA or ALT levels.

 At the end of the open label phase (Week 48), the mean change from baseline HBV DNA by PCR for patients originally assigned to entecavir was -4.20 log10 copies/mL (n = 43); 4/51 (8%) patients had HBV DNA < 300 copies/mL by PCR; and 13/35 (37%) patients with abnormal ALT at baseline had ALT normalisation. Entecavir has not been evaluated in HIV/HBV co-infected patients who are not concurrently receiving effective HIV treatment (see Section 4.4 Special Warnings and Precautions for Use, Co-infection with HIV).
At the end of the open label phase (Week 48), the mean change from baseline HBV DNA by PCR for patients originally assigned to entecavir was -4.20 log10 copies/mL (n = 43); 4/51 (8%) patients had HBV DNA < 300 copies/mL by PCR; and 13/35 (37%) patients with abnormal ALT at baseline had ALT normalisation. Entecavir has not been evaluated in HIV/HBV co-infected patients who are not concurrently receiving effective HIV treatment (see Section 4.4 Special Warnings and Precautions for Use, Co-infection with HIV). Dosage adjustment is recommended for patients with a creatinine clearance < 50 mL/min, including patients on hemodialysis or CAPD. (See Section 4.2 Dose and Method of Administration, Renal impairment.)
Dosage adjustment is recommended for patients with a creatinine clearance < 50 mL/min, including patients on hemodialysis or CAPD. (See Section 4.2 Dose and Method of Administration, Renal impairment.)
