What is in this leaflet
This leaflet answers some common questions about BAVENCIO.
It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have benefits and risks. Your doctor has weighed the risks of treating you with BAVENCIO against the benefits expected for you.
If you have any concerns about receiving this medicine, talk to your doctor, nurse or the hospital pharmacist.
Keep this leaflet in a safe place. You may need to read it again.
What BAVENCIO is used for
BAVENCIO contains the active substance avelumab, a monoclonal antibody, which is a protein designed to recognise and attach to a specific target in the body to help the immune system attack and destroy cancer cells.
BAVENCIO is used to treat:
- a rare type of skin cancer called metastatic Merkel cell carcinoma (mMCC) where the disease has spread.
- a type of cancer in the bladder or urinary tract called urothelial carcinoma, when the disease is advanced or metastatic but has not progressed with platinum-based chemotherapy.
- a type of kidney cancer called renal cell carcinoma (RCC), when it is advanced, in combination with another medicine, axitinib. It is important that you also read the Consumer Medicine Information for axitinib. If you have any questions about axitinib, ask your doctor.
This medicine is available only with a doctor's prescription.
Ask your doctor if you have any questions about why BAVENCIO has been prescribed for you. Your doctor may have prescribed it for another reason.
Before you are given BAVENCIO
When you must not be given it
Do not use BAVENCIO:
- if you are allergic (hypersensitive) to avelumab or any of the other ingredients of BAVENCIO. If you are not sure, talk to your doctor.
Some of the symptoms of an allergic reaction may include:
- shortness of breath, wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
Do not use BAVENCIO after the expiry date printed on the pack. The expiry date refers to the last day of that month. If you take it after the expiry date has passed, it may not work well. Your pharmacist or nurse will check the expiry date on the packaging.
Do not use BAVENCIO if the packaging is torn or shows signs of tampering. Your pharmacist or nurse will check the packaging for any signs of damage.
If you are not sure whether you should start using this medicine, talk to your doctor.
Before you are given BAVENCIO
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have, or have had, any of the following medical conditions:
- an autoimmune disease (a condition where the body attacks its own cells) like Crohn's, Guillain-Barré, ulcerative colitis or lupus. If you already have an autoimmune disease, your risk of immune-mediated side effects may be higher as BAVENCIO may cause inflammation in parts of your body. You may also experience frequent flares of your autoimmune disease, which in the majority of cases are mild.
- human immunodeficiency virus (HIV) infection or acquired immune deficiency syndrome (AIDS)
- hepatitis B or hepatitis C
- taking any medicines that suppress your immune system
- have had an organ transplant.
If you have not told your doctor about any of the above, tell them before you are given BAVENCIO.
Once you are treated with BAVENCIO, your doctor may give you corticosteroids to reduce any possible side effects that you may have during your treatment.
Talk to your doctor or nurse before receiving BAVENCIO as it may cause:
- problems with your lungs such as breathing difficulties, or cough. These may be signs of inflammation of the lungs (pneumonitis).
- problems with your liver (hepatitis). Signs and symptoms of hepatitis may include yellowing of the whites of the eyes or skin (jaundice), severe nausea or vomiting, pain on the right side of your stomach area, drowsiness, dark urine (tea coloured), bleeding or bruising more easily than normal, tiredness, abnormal liver function tests.
- problems with your intestines (colitis) such as diarrhoea (loose stools), more bowel movements than usual, blood in your stools or dark, tarry, sticky stools, severe stomach area (abdomen) pain or tenderness.
- problems with your pancreas (pancreatitis). Signs of pancreatitis may include abdominal pain, nausea and vomiting.
- problems with your hormone producing glands (including the thyroid, pituitary, and adrenal glands) that may affect how these glands work. Signs and symptoms that your glands are not working properly may include rapid heart beat, increased sweating, extreme tiredness, weight changes, changes in mood or behaviour, such as irritability or forgetfulness, feeling cold, very low blood pressure (dizziness or fainting, fatigue, nausea), headache.
- problems with your pancreas (Type 1 diabetes), including a serious, sometimes life-threatening problem due to increased acid in the blood produced from diabetes (diabetic ketoacidosis). Signs and symptoms include feeling more hungry or thirsty than usual, needing to urinate more often, weight loss, feeling tired, having difficulty thinking clearly, breath that smells sweet or fruity, feeling sick or being sick, stomach pain, deep or fast breathing.
- problems with your kidneys such as abnormal kidney function tests, need to urinate less than usual, blood in your urine, swelling in your ankles.
- problems with your muscles (myositis, myasthenia gravis/ myasthenic syndrome, polymyalgia rheumatica) such as muscle pain, stiffness or weakness.
- problems with your bile ducts (sclerosing cholangitis) such as pain in the upper right part of the stomach, swelling of the liver or spleen, fatigue, itching, or yellowing of the skin or the whites of eyes.
- problems with your joints (arthritis) such as joint pain, stiffness, and swelling.
- problems with your glands that make moisture for the body (Sjogren's syndrome) such as tears and saliva leading to dry eyes and dry mouth.
- problems with your heart:
- (myocarditis) such as trouble breathing, dizziness or fainting, fever, chest pain, chest tightness or flu-like symptoms.
- (major adverse cardiovascular events) such as trouble breathing, chest discomfort, or swelling of lower legs or hands. - infusion-related reactions, which may include chills or shaking, hives, shortness of breath or wheezing, fever, back pain and/or abdominal pain, bumpy rash or skin wheals, flushing, low blood pressure (dizziness, fatigue, nausea).
In addition, autoimmune haemolytic anaemia (headaches, short of breath, looking pale, and yellowing of the skin and/or eyes) and systemic inflammatory response syndrome (redness and swelling in the affected parts of your body, pain, loss of function of parts of your body, fatigue, fast heart rate, abnormal breathing, fever or low body temperature, shaking or chills, warm or clammy/sweaty skin, rash, confusion, agitation or other mental changes, loss of consciousness) have been reported with BAVENCIO or other products in this class (PD-1/ PD-L1 blocking antibodies).
Should any of the above occur, your doctor may:
- give you other medicines in order to prevent complications and reduce your symptoms
- withhold the next dose of BAVENCIO
- or stop your treatment with BAVENCIO altogether.
Pregnancy
Tell your doctor if you are pregnant or plan to become pregnant. BAVENCIO must not be used during pregnancy as it may cause harm to your unborn baby.
Make sure you maintain adequate contraception during treatment and for at least one month after your last dose.
Breast-feeding
Tell your doctor if you are breastfeeding or wish to breastfeed. It is unknown if BAVENCIO is passed into your breast milk. A risk to the breast-fed infant cannot be excluded.
You should not breastfeed during treatment and for at least one month after your last dose.
Use in Children
The effectiveness of BAVENCIO in children under the age of 12 years has not been established.
Taking other medicines
Tell your doctor, pharmacist or nurse if you are taking or have recently taken any other medicines, including medicines obtained without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and BAVENCIO may interfere with each other. These include:
- medicines that make your immune system weak, such as steroids.
These medicines may be affected by BAVENCIO or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor or nurse will have more information on medicines to be careful with or to avoid while using BAVENCIO.
How to use BAVENCIO
Follow all directions given to you by your doctor, nurse or pharmacist carefully. They may differ from the information contained in this leaflet.
A doctor experienced in the use of medicines for cancer will supervise your treatment with BAVENCIO.
You will receive BAVENCIO as an infusion (a drip) into a vein (intravenously) over a period of 60 minutes, once every 2 weeks.
Your doctor will decide how many treatments you need.
You will also receive premedication (paracetamol and an antihistamine) prior to the first four infusions of BAVENCIO, and prior to further infusions of BAVENCIO if your doctors believes this is necessary. These premedications are given to help minimise potential reactions to the BAVENCIO infusion.
How much to use
BAVENCIO is a concentrated solution for infusion. The recommended dose is either 10 mg per kg of your body weight or 800 mg of BAVENCIO. Your doctor will calculate the correct dose and dilution for you.
BAVENCIO has to be diluted with 0.9% or 0.45% saline solution before use. More than one vial is required to obtain the required dose.
How long to use BAVENCIO for
Your doctor will decide how long you will receive BAVENCIO based on your response to the medicine and the type of cancer you have.
If you miss a dose
It is very important for you to keep all your appointments to receive BAVENCIO.
If you miss a dose, talk to your doctor or nurse and arrange another visit as soon as possible.
If you take too much (overdose)
Immediately contact your doctor or the Poisons Information Centre (telephone 13 11 26) for advice, or go to Accident and Emergency at the nearest hospital, if you think that you or anyone else may have taken too much BAVENCIO.
Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
While you are being treated with BAVENCIO
Things you must do
Limit your exposure to sunlight by wearing hat, protective clothing and sunscreen when you go outside.
Keep all appointments with your doctor so that your progress can be checked. Your doctor may do blood tests or other laboratory tests from time to time to prevent unwanted side effects during your treatment with BAVENCIO.
Before starting any new medicine, remind your doctor or pharmacist that you are receiving BAVENCIO.
Tell all the doctors, dentists and pharmacists who are treating you that you are receiving BAVENCIO.
If you become pregnant while you are treated with BAVENCIO, tell your doctor immediately.
Things to be careful of
Be careful driving or operating machinery until you know how BAVENCIO affects you. Treatment-related symptoms can affect your concentration and ability to react.
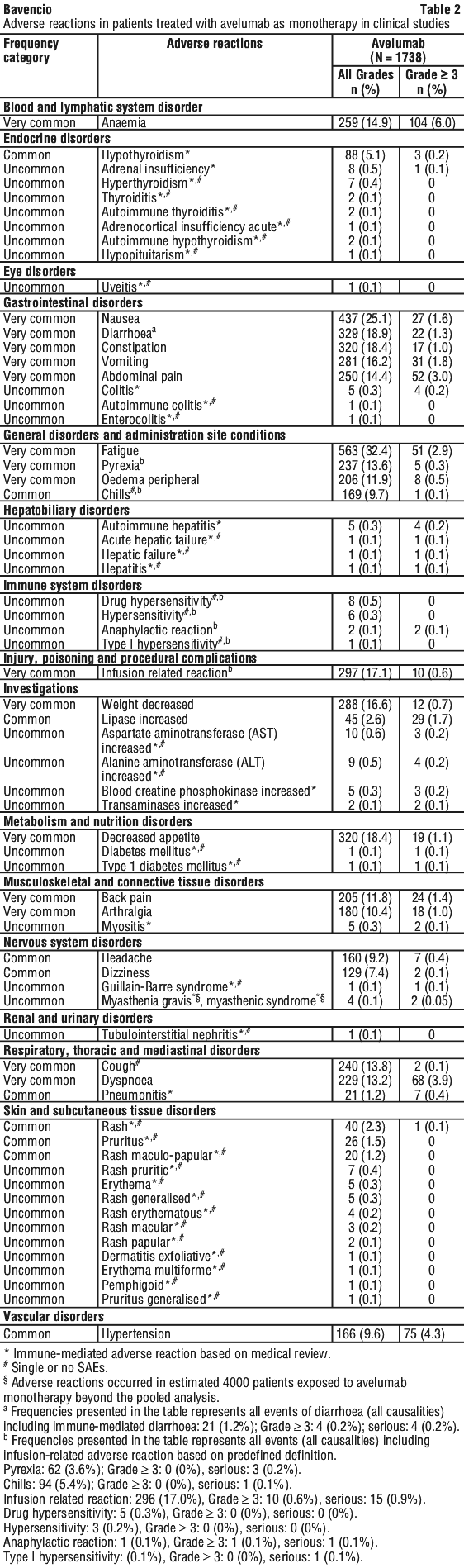
Side effects
Tell your doctor or nurse as soon as possible if you do not feel well while you are receiving BAVENCIO.
All medicines, including BAVENCIO, can have unwanted side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Be aware of important symptoms of inflammation. BAVENCIO acts on your immune system and may cause inflammation in parts of your body. Inflammation may cause serious damage to your body and some inflammatory conditions may be life-threatening and need treatment or withdrawal of BAVENCIO.
Do not try to treat your symptoms with other medicines.
Do not be alarmed by this list of possible side effects. You may not experience any of them.
Ask your doctor to answer any questions you may have.
Tell your doctor or nurse if you notice any of the following, particularly if they worry you:
- feeling tired or weak
- muscle or back pain, stomach area pain, joint pain
- loose stools, vomiting, nausea, constipation
- swelling in the arms, feet or legs
- feeling less hungry, weight loss
- cough
- rash, itching, redness of the skin
- fever, chills
- shortness of breath
- dizziness, headache
- high blood pressure
- low blood pressure
- flushing
- difficulty in speaking or breathing, hoarseness
- urinary tract infection (UTI) such as burning sensation when urinating, strong or frequent or urge to urinate
The above list includes the common side effects of BAVENCIO.
If any of the following happen, tell your doctor immediately or go to Accident and Emergency at your nearest hospital:
- signs of problems with your liver: yellowing of the whites of the eyes or skin, severe nausea or vomiting, pain on the right side of your stomach area, drowsiness, dark urine (tea coloured), bleeding or bruising more easily than normal, feeling less hungry than usual, tiredness or abnormal liver function tests
- signs of problems with your thyroid, adrenal or pituitary glands (hormone producing glands): extreme tiredness, rapid heartbeat, increased sweating, weight changes, changes in mood or behaviour, such as irritability or forgetfulness, feeling cold, very low blood pressure (fainting, dizziness, fatigue, nausea), headache
- signs of problem with your intestines: diarrhoea, more bowel movements than usual, blood in your stools or dark, tarry, sticky stools, severe stomach pain or tenderness
- signs of problems with your lungs: breathing difficulties, coughing
- signs of problems with your pancreas (type 1 diabetes including a serious, sometimes life-threatening problem due to increase acid in the blood produced from diabetes (diabetic ketoacidosis)) may include feeling more hungry or thirsty than usual, needing to urinate more often, weight loss, and feeling tired, having difficulty thinking clearly, breath that smells sweet or fruity, feeling sick or being sick, stomach pain, deep or fast breathing
- signs of inflammation of the kidney may include abnormal kidney function tests, needing to urinate less than usual, blood in your urine, or swelling in your ankles
- signs of problems with your heart: trouble breathing, dizziness or fainting, fever, chest pain, chest tightness, swelling in lower legs or hands, or flu-like symptoms
- signs of problems with your muscles: muscle pain, stiffness or weakness
- signs of problems with your bile ducts: pain in the upper right part of the stomach, swelling of the liver or spleen, fatigue, itching, or yellowing of the skin or the whites of eyes
- signs of problems with your joints: joint pain, stiffness, swelling
- signs of problems with your glands that make moisture for the body, such as tears and saliva: dry eyes, dry mouth
- signs of problems with your pancreas: abdominal pain, nausea, vomiting
- signs of problems with your eyes: eye pain, eye redness, sensitivity to light, blurred or cloudy vision, small shapes moving across your field of vision (floaters), loss of peripheral vision (the ability to see objects at the side of your field of vision)
- Guillain-Barré Syndrome (an immune system disorder that causes nerve inflammation and can result in pain, numbness, muscle weakness and difficulty walking)
- signs of an allergic reaction (anaphylactic reaction and/or hypersensitivity): shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body; rash, itching or hives on the skin
The above list includes serious side effects. You may need urgent medical attention or hospitalisation.
Please note that these signs and symptoms are sometimes delayed, and may develop weeks or months after your last dose.
Infusion-related side effects
Tell your doctor or nurse if you experience any of the following symptoms, during or after treatment with BAVENCIO:
- fever
- back and/or abdominal pain
- chills or shaking
- shortness of breath or wheezing
- bumpy rash or skin wheals
- flushing
- low blood pressure (dizziness, fatigue, nausea).
Sometimes these symptoms may occur up to several hours later. To recognise early signs of these side effects, you will be monitored closely while you are receiving each infusion.
Your doctor may consider reducing the infusion rate, stop the infusion temporarily or permanently in order to manage these symptoms.
Tell your doctor if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some patients.
After being treated with BAVENCIO
Storage
Undiluted BAVENCIO must be kept in a refrigerator at 2° to 8°C, but it must not be frozen. Store in the original package in order to protect from light.
Each vial is intended for single use only. When the needed amount of solution has been withdrawn from the vial, any remaining concentrated solution must be discarded.
Product description
What it looks like
BAVENCIO is a clear, colourless, to slightly yellow solution. It is supplied in 10 mL colourless glass vials with a rubber stopper and aluminium seal. Each pack contains 1 vial.
Ingredients
The active ingredient in BAVENCIO is avelumab. Each 10 mL vial contains 200 mg avelumab.
The solution also contains the following inactive ingredients:
- mannitol
- glacial acetic acid
- polysorbate 20
- sodium hydroxide
- water for injections.
BAVENCIO solution does not contain any preservative.
Sponsor
Merck Healthcare Pty Ltd
Suite 1, Level 1
Building B
11 Talavera Rd
Macquarie Park NSW 2113
Australia
Merck Medical Information: 1800 633 463
Australian registration number
AUST R 282729
Date of preparation
July 2024
® Registered Trademark of Merck KGaA, Darmstadt, Germany
a010-0724
Published by MIMS August 2024


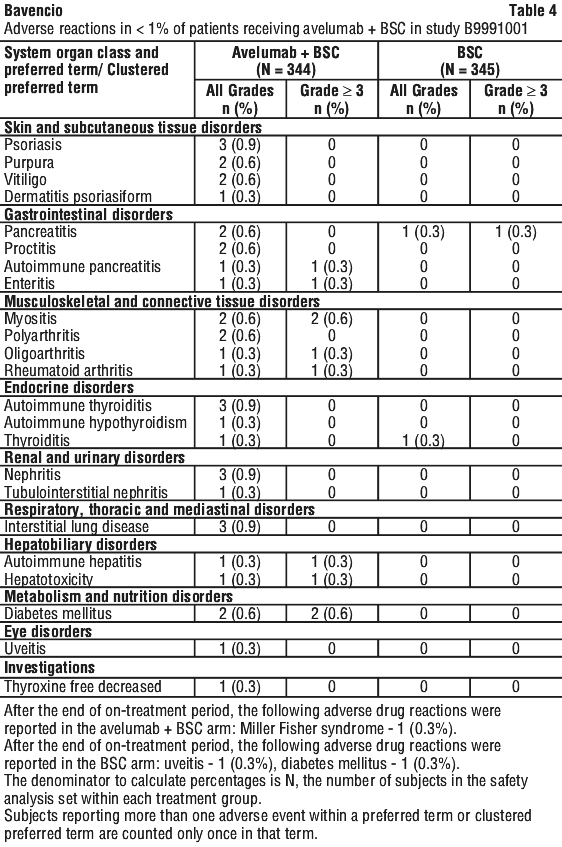

 Drug eruption was reported in one patient receiving avelumab in combination with axitinib in study B9991002.
Drug eruption was reported in one patient receiving avelumab in combination with axitinib in study B9991002.



 In addition, a statistically significant improvement in OS was demonstrated in patients with PD-L1 positive tumours for Bavencio plus BSC compared to BSC alone (HR 0.56; 95% CI: 0.40, 0.79; 2 sided p-value 0.0007). The median OS was not reached (95% CI: 20.3 months, not reached) in the Bavencio plus BSC arm, and 17.1 months (95% CI: 13.5, 23.7) in the BSC alone arm. Consistent results were observed across pre-specified subgroups, including best response to first line induction chemotherapy, and sites of metastasis as shown in Figure 3.
In addition, a statistically significant improvement in OS was demonstrated in patients with PD-L1 positive tumours for Bavencio plus BSC compared to BSC alone (HR 0.56; 95% CI: 0.40, 0.79; 2 sided p-value 0.0007). The median OS was not reached (95% CI: 20.3 months, not reached) in the Bavencio plus BSC arm, and 17.1 months (95% CI: 13.5, 23.7) in the BSC alone arm. Consistent results were observed across pre-specified subgroups, including best response to first line induction chemotherapy, and sites of metastasis as shown in Figure 3.
 At the time of the first interim analysis, the OS data were still immature. The observed HR was 0.78 (95% CI: 0.554, 1.084) in favour of avelumab in combination with axitinib. Median OS was not yet reached in either treatment arms. The probability of being alive at 12 months was 86.3% (95%CI: 82.2%, 89.5%) for the avelumab plus axitinib arm and 83.0% (95% CI: 78.8%, 86.5%) for the sunitinib arm.
At the time of the first interim analysis, the OS data were still immature. The observed HR was 0.78 (95% CI: 0.554, 1.084) in favour of avelumab in combination with axitinib. Median OS was not yet reached in either treatment arms. The probability of being alive at 12 months was 86.3% (95%CI: 82.2%, 89.5%) for the avelumab plus axitinib arm and 83.0% (95% CI: 78.8%, 86.5%) for the sunitinib arm. A statistically significant improvement in PFS was observed in both patients with PD-L1-positive tumours and all patients irrespective of PD-L1 expression who received the combination of avelumab with axitinib, with a 39% and 31% reduction of the risk of progression or death as compared to patients treated with sunitinib, respectively. Improvement of PFS was observed across all risk groups based on IMDC and MSKCC.
A statistically significant improvement in PFS was observed in both patients with PD-L1-positive tumours and all patients irrespective of PD-L1 expression who received the combination of avelumab with axitinib, with a 39% and 31% reduction of the risk of progression or death as compared to patients treated with sunitinib, respectively. Improvement of PFS was observed across all risk groups based on IMDC and MSKCC. Avelumab is a human monoclonal IgG1 antibody directed against the immunomodulatory cell surface ligand protein PD-L1 and produced in Chinese hamster ovary cells by recombinant DNA technology.
Avelumab is a human monoclonal IgG1 antibody directed against the immunomodulatory cell surface ligand protein PD-L1 and produced in Chinese hamster ovary cells by recombinant DNA technology.