1 Name of Medicine
Biktarvy (bictegravir (as sodium)/emtricitabine/tenofovir alafenamide).
2 Qualitative and Quantitative Composition
Each film-coated tablet contains bictegravir sodium equivalent to 50 mg of bictegravir (BIC), 200 mg of emtricitabine (FTC), and tenofovir alafenamide (TAF) fumarate equivalent to 25 mg of tenofovir alafenamide.
For the full list of excipients, see Section 6.1 List of Excipients.
3 Pharmaceutical Form
The tablets are film-coated, capsule shaped and purplish-brown in colour. Each tablet is debossed with 'GSI' on one side and the number "9883" on the other side.
4.1 Therapeutic Indications
Biktarvy is indicated for the treatment of HIV-1 infection in adults and paediatric patients weighing at least 25 kg who are antiretroviral therapy (ART)-naïve or to replace the current antiretroviral regimen in those who are virologically-suppressed (HIV-1 RNA < 50 copies/mL) on a stable antiretroviral regimen at the start of therapy with no history of treatment failure, and no known substitutions associated with resistance to bictegravir or tenofovir.
4.2 Dose and Method of Administration
In adults and paediatric patients weighing at least 25 kg, the dose of Biktarvy is one tablet taken orally once daily with or without food.
No data are available on which to make a dose recommendation for paediatric patients weighing less than 25 kg.
Elderly.
No dose adjustment is required for elderly patients.
Renal impairment.
No dose adjustment of Biktarvy is required in adult patients with estimated creatinine clearance ≥ 30 mL/min or in adult patients with end stage renal disease (ESRD; estimated creatinine clearance < 15 mL/min) who are receiving chronic haemodialysis. On days of haemodialysis, administer the daily dose of Biktarvy after completion of haemodialysis treatment.
Initiation of Biktarvy is not recommended in patients with estimated creatinine clearance ≥ 15 and < 30 mL/min, or < 15 mL/min who are not receiving chronic haemodialysis, as the safety of Biktarvy has not been established in these populations.
No data are available to make a dose recommendation in paediatric patients with renal impairment.
Hepatic impairment.
No dose adjustment of Biktarvy is required in patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment. Biktarvy has not been studied in patients with severe hepatic impairment (Child-Pugh Class C) (see Section 5.2 Pharmacokinetic Properties, Pharmacokinetics in special populations).4.3 Contraindications
Biktarvy is contraindicated in patients with known hypersensitivity to BIC, FTC, TAF or to any of the excipients.
Coadministration with dofetilide is contraindicated due to the potential for increased dofetilide plasma concentrations and associated serious and/or life-threatening events.
Coadministration with rifampicin is contraindicated due to decreased BIC plasma concentrations, which may result in the loss of therapeutic effect and development of resistance to Biktarvy.
Please see Table 4 for Established and other potentially significant drug interactions. In addition, prescribing information for any drug coadministered with Biktarvy should be consulted to exclude significant interaction or contraindication.
4.4 Special Warnings and Precautions for Use
General.
Patients receiving Biktarvy or any other antiretroviral therapy may continue to develop opportunistic infections and other complications of HIV infection, and therefore should remain under close clinical observation by physicians experienced in the treatment of patients with HIV associated diseases.
While effective viral suppression with antiretroviral therapy has been proven to substantially reduce the risk of HIV transmission, a residual risk cannot be excluded. Appropriate precautions must continue to be used. Patients should also be informed that Biktarvy is not a cure for HIV infection.
HIV and hepatitis B virus (HBV) co-infection.
Discontinuation of Biktarvy therapy in patients co-infected with HIV-1 and HBV may be associated with severe acute exacerbations of hepatitis due to the FTC and TAF components of Biktarvy. Patients co-infected with HIV-1 and HBV who discontinue Biktarvy should be closely monitored with both clinical and laboratory follow-up for at least several months after stopping treatment. If appropriate, anti-hepatitis B therapy may be warranted, especially in HBV co-infected patients with advanced liver disease or cirrhosis since post-treatment exacerbation of hepatitis may lead to hepatic decompensation.
Use with other antiretroviral products.
Biktarvy should not be coadministered with products containing any of the same components, BIC, TAF or FTC; or with products containing lamivudine or tenofovir disoproxil fumarate (TDF). Biktarvy should not be administered with adefovir dipivoxil.
Immune reconstitution syndrome.
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including FTC, a component of Biktarvy. In HIV-infected patients with severe immune deficiency at the time of initiation of antiretroviral therapy, an inflammatory reaction to asymptomatic or residual opportunistic pathogens may arise and cause serious clinical conditions, or aggravation of symptoms. Typically, such reactions have been observed within the first few weeks or months of initiation of antiretroviral therapy. Relevant examples include cytomegalovirus retinitis, generalised and/or focal mycobacterial infections, HBV and Pneumocystis jirovecii pneumonia. Any inflammatory symptoms should be evaluated and treatment instituted when necessary.
Autoimmune disorders (such as autoimmune hepatitis) have also been reported to occur in the setting of immune reconstitution; however, the reported time to onset is more variable, and these events can occur many months after initiation of treatment.
Paediatric use.
No dose adjustment is required for paediatric patients weighing at least 25 kg. No data are available on which to make a dose recommendation for paediatric patients weighing less than 25 kg.
Use in the elderly.
No dose adjustment of Biktarvy is required for elderly patients. Clinical trials included 111 patients aged 65 years and over who received Biktarvy. No differences in safety or efficacy have been observed between elderly patients and those less than 65 years of age.
Use in renal impairment.
Post marketing cases of renal impairment, including acute renal failure, proximal renal tubulopathy (PRT), and Fanconi syndrome have been reported with tenofovir alafenamide containing products; while most of these cases were characterised by potential confounders that may have contributed to the reported renal events, it is also possible these factors may have predisposed patients to tenofovir-related adverse events.
Patients taking tenofovir prodrugs who have impaired renal function and those taking nephrotoxic agents, including non-steroidal anti-inflammatory drugs, are at increased risk of developing renal-related adverse reactions.
Prior to or when initiating Biktarvy, and during treatment with Biktarvy on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus. Discontinue Biktarvy in patients who develop clinically significant decreases in renal function or evidence of Fanconi syndrome.
No dose adjustment of Biktarvy is required in adult patients with estimated creatinine clearance greater than or equal to 30 mL/min.
The safety, virologic, and immunologic responses of FTC+TAF was evaluated through 144 weeks in an open-label clinical study GS-US-292-0112 (Study 0112) in which 248 HIV-1 infected adult patients who were either treatment-naïve (N=6) or virologically suppressed (N=242) with mild to moderate renal impairment (eGFR by Cockcroft-Gault method 30-69 mL/min) received FTC+TAF in combination with EVG+COBI as a fixed-dose combination tablet. The safety profile of FTC+TAF in patients with mild to moderate renal impairment was similar to safety data that from patients with normal renal function.
The safety of FTC + TAF (components of Biktarvy) was evaluated in a single arm, open-label clinical study (GS-US-292-1825 ["Study 1825"]), in which 55 virologically-suppressed HIV-1 infected patients with ESRD (eGFR by Cockcroft-Gault method < 15 mL/min) on chronic haemodialysis received FTC+TAF in combination with EVG+COBI as a fixed-dose combination tablet for 96 weeks. In an extension phase of Study 1825, 10 patients switched to Biktarvy for 48 weeks. The safety profile of FTC+TAF in patients with ESRD on chronic haemodialysis was similar to that in patients with normal renal function, and no additional adverse reactions were identified in patients administered Biktarvy in this study.
Initiation of Biktarvy is not recommended in patients with estimated creatinine clearance ≥ 15 and < 30 mL/min, or < 15 mL/min who are not receiving chronic haemodialysis, as the safety of Biktarvy has not been established in these populations. (see Section 4.2 Dose and Method of Administration).
Use in hepatic impairment.
No dose adjustment of Biktarvy is required in patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment. Biktarvy has not been studied in patients with severe hepatic impairment (Child-Pugh Class C) (see Section 5.1, Clinical trials; Section 5.2 Pharmacokinetic Properties).
Lactic acidosis/ severe hepatomegaly with steatosis.
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogs, including FTC, a component of Biktarvy, and tenofovir DF, another prodrug of tenofovir, alone or in combination with other antiretrovirals. Treatment with Biktarvy should be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
Co-administration of other medicinal products.
Under fasted conditions, Biktarvy should not be co-administered simultaneously with magnesium/aluminium-containing antacids due to the expected substantial decrease of BIC exposure.
Under fasted conditions, Biktarvy should be administered at least 2 hours before or 6 hours after taking antacids containing magnesium and/or aluminium (see Section 4.5).
If taken together with food, Biktarvy and magnesium/aluminium-containing antacids can be co-administered simultaneously (see Section 4.5).
Effects on laboratory tests.
No data available.4.5 Interactions with Other Medicines and Other Forms of Interactions
General.
As Biktarvy contains BIC, FTC and TAF, any interactions that have been identified with these agents individually may occur with Biktarvy.
Bictegravir.
BIC inhibits organic cation transporter 2 (OCT2) and multidrug and toxin extrusion transporter 1 (MATE1) in vitro. Coadministration of Biktarvy with the OCT2 and MATE1 substrate metformin did not result in a clinically significant increase in metformin exposure. Biktarvy may be coadministered with substrates of OCT2 and MATE1 except dofetilide, which is contraindicated due to the potential for increased dofetilide plasma concentrations and associated serious and/or life-threatening events (see Section 4.3 Contraindications).
BIC is a substrate of P-gp and BCRP in vitro. However, clinical drug interaction data show that P-gp/BCRP does not play a clinically significant role in the disposition of BIC.
BIC is primarily eliminated through hepatic metabolism by CYP3A and UGT1A1. Drugs that are potent inducers of both CYP3A and UGT1A1, such as rifampicin, may significantly decrease plasma exposures of BIC leading to reduced therapeutic effect of BIC. Coadministration with rifampicin is contraindicated. Potent inhibitors of both CYP3A and UGT1A1, such as atazanavir, may significantly increase BIC exposure, and coadministration is not recommended.
BIC is not an inhibitor or inducer of CYP3A in vivo.
Emtricitabine.
In vitro and clinical pharmacokinetic drug-drug interaction studies have shown that the potential for CYP-mediated interactions involving FTC with other medicinal products is low. FTC is primarily excreted by the kidneys by a combination of glomerular filtration and active tubular secretion. No drug-drug interactions due to competition for renal excretion have been observed; however, coadministration of FTC with drugs that are eliminated by active tubular secretion may increase concentrations of FTC, and/or the coadministered drug.
Drugs that decrease renal function may increase concentrations of FTC.
In drug interaction studies conducted with FTC and with TDF, coadministration of FTC and famciclovir had no effect on the Cmax or AUC of either drug.
Tenofovir alafenamide.
TAF is a substrate of P-glycoprotein (P-gp) and BCRP. Drugs that strongly affect P-gp and BCRP activity may lead to changes in TAF absorption. TAF is not an inhibitor or inducer of CYP3A in vivo.
Drug interaction studies.
Drug-drug interaction studies were conducted with Biktarvy or various combinations of the components of Biktarvy (BIC, FTC or TAF).
The effects of coadministered drugs on the exposure of BIC are shown in Table 1. The effects of coadministered drugs on the exposure of TAF are shown in Table 2. The effects of BIC and/or TAF on the exposure of coadministered drugs are shown in Table 3.


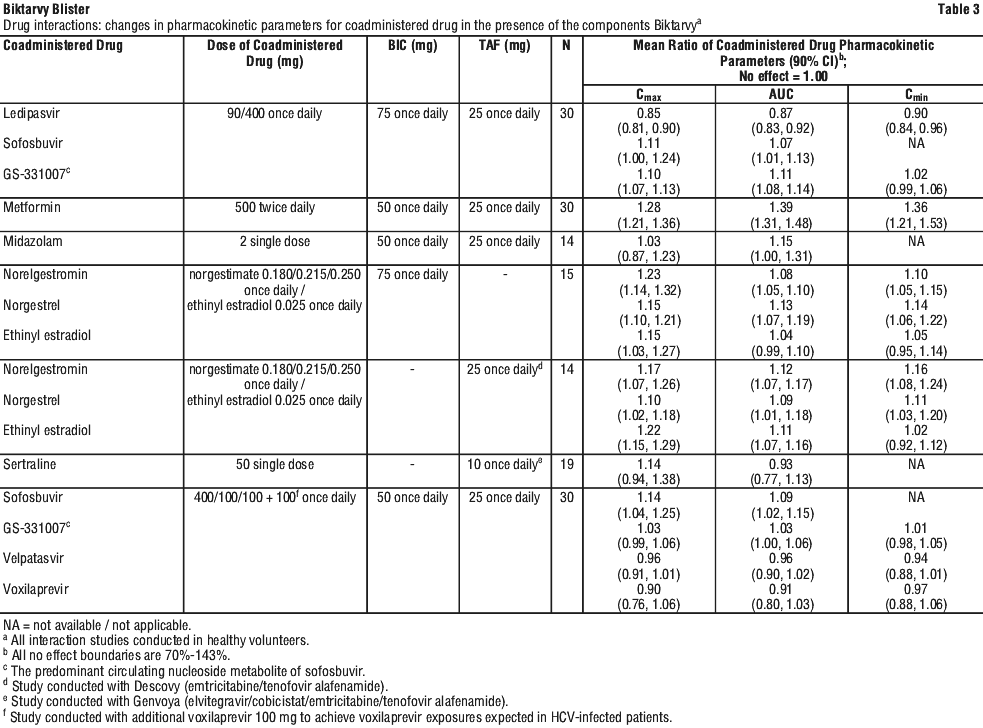
Effects of concomitant drugs on the pharmacokinetics of Biktarvy.
BIC, a component of Biktarvy, is a substrate of CYP3A and UGT1A1. Coadministration of BIC and drugs that potently induce both CYP3A and UGT1A1 may significantly decrease plasma concentrations of BIC, which may result in loss of therapeutic effect of Biktarvy and development of resistance. Coadministration of BIC with drugs that potently inhibit both CYP3A and UGT1A1 may significantly increase plasma concentrations of BIC.
TAF, a component of Biktarvy, is transported by P-glycoprotein (P-gp) and BCRP. Drugs that strongly affect P-gp and BCRP activity may lead to changes in TAF absorption (see Table 4). Drugs that induce P-gp activity are expected to decrease the absorption of TAF, resulting in decreased plasma concentration of TAF, which may lead to loss of therapeutic effect of Biktarvy and development of resistance. Coadministration of Biktarvy with other drugs that inhibit P-gp and BCRP may increase the absorption and plasma concentration of TAF.
Established and other potentially significant interactions.
Biktarvy is a complete regimen for the treatment of HIV-1 infection. Therefore, comprehensive information regarding drug-drug interactions with other antiretroviral products is not provided.
Drug interaction information for Biktarvy with potential concomitant drugs is summarised in Table 4. The drug interactions described are based on studies conducted with Biktarvy or the components of Biktarvy (BIC, FTC, and TAF) as individual agents, or are potential drug interactions that may occur with Biktarvy.
The table is not all-inclusive (see Section 4.3 Contraindications).

Drugs without clinically significant interactions with Biktarvy.
Based on drug interaction studies conducted with Biktarvy or the components of Biktarvy, no clinically significant drug interactions were observed or are expected with: amlodipine, atorvastatin, buprenorphine, drospirenone, ethinyl estradiol, famotidine, fluticasone, itraconazole, ketoconazole, ledipasvir/sofosbuvir, metformin, methadone, midazolam, naloxone, norbuprenorphine, norgestimate, omeprazole, sertraline, sofosbuvir, sofosbuvir/velpatasvir, or sofosbuvir/velpatasvir/voxilaprevir.4.6 Fertility, Pregnancy and Lactation
Effects on fertility.
No reproductive toxicity studies have been conducted with BIC, FTC and TAF in combination.
Bictegravir.
BIC did not affect fertility, reproductive performance or embryonic viability in male and female rats at 29 times higher exposures (AUC) than in humans at the recommended dose of 50 mg BIC in Biktarvy.
Emtricitabine.
FTC did not affect fertility in male rats at approximately 140 times or in male and female mice at approximately 60 times higher exposures (AUC) than in humans given the recommended 200 mg daily dose in Biktarvy. Fertility was normal in the offspring of mice exposed daily from before birth (in utero) through sexual maturity at daily exposures (AUC) of approximately 60 times higher than human exposures at the recommended 200 mg daily dose in Biktarvy.
Tenofovir alafenamide.
There were no effects on fertility, mating performance or early embryonic development when TAF was administered to male rats at a dose equivalent to 155 times (25 mg TAF) the human dose based on body surface area comparisons for 28 days prior to mating and to female rats for 14 days prior to mating through day seven of gestation.
(Category B3)
Biktarvy was evaluated in a clinical study of 33 HIV-1 infected virologically suppressed (HIV-1 RNA < 50 copies/mL) pregnant adults administered 50 mg/200 mg/25 mg Biktarvy once daily from the second or third trimester through postpartum. Exposures of BIC, FTC, and TAF were lower during pregnancy as compared to postpartum; these decreases are not considered to be clinically relevant based on exposure-response relationships; however, specific dosage adjustments for coadministered oral medications or supplements containing polyvalent cations are recommended in pregnant patients (see Section 4.5; Section 5.2).
All 32 adult participants who completed the study maintained viral suppression (HIV-1 RNA < 50 copies/mL) during pregnancy, at delivery, and through Week 18 postpartum. The median (Q1, Q3) CD4+ cell count at baseline was 558 (409, 720) cells/microL, and the median (Q1, Q3) change in CD4+ cell count from baseline to Week 12 postpartum was 159 (27, 296) cells/microL. All 29 neonate participants had negative/nondetectable HIV-1 PCR results at 4 to 8 weeks post-birth. Biktarvy was well tolerated during pregnancy and postpartum; there were no new safety findings compared to the known safety profile of Biktarvy in HIV-1 infected adults.
Biktarvy may be used during pregnancy if the potential benefit justifies the potential risk to the fetus.
Bictegravir.
Studies in animals have shown no evidence of teratogenicity or an effect on reproductive function. In offspring from rat and rabbit dams treated with BIC during pregnancy, there were no toxicologically significant effects on developmental endpoints 36 times (rat) or 1.4 times (rabbit) higher exposures (AUC) than in humans at the recommended dose of 50 mg BIC in Biktarvy.
Emtricitabine.
No evidence of embryofoetal toxicity or teratogenicity was observed in mice or rabbits at respective FTC exposures (AUC) of 50 and 130 fold the clinical exposure. Impaired weight gain observed in pregnant rabbits at doses resulting in FTC exposures (AUC) at least 33 times the clinical exposure was not associated with any adverse fetal effects.
Tenofovir alafenamide.
Embryofetal development studies performed in rats and rabbits revealed no evidence of embryolethality, fetotoxicity or teratogenicity due to TAF. The embryo-fetal NOAELs in rats and rabbits occurred at TAF exposures (AUC) similar to and 53 times higher than, respectively, the exposure in humans at the recommended daily dose.
In animal studies, BIC was detected in the plasma of nursing rat pups likely due to the presence of BIC in milk, without effects on nursing pups at maternal exposures 30 times higher exposures (AUC) than in humans at the recommended dose of 50 mg BIC in Biktarvy. In animal studies it has been shown that tenofovir is secreted into milk. It is not known whether BIC or TAF is secreted in human milk. In humans, samples of breast milk obtained from five HIV-1 infected mothers given Truvada (TDF/FTC) show that FTC is secreted in human milk at estimated neonatal concentrations 3 to 12 times higher than the FTC IC50 but 3 to 12 times lower than the Cmin achieved from oral administration of FTC. Breastfeeding infants whose mothers are being treated with FTC may be at risk for developing viral resistance to FTC. Other FTC-associated risks in infants breastfed by mothers being treated with FTC are unknown.
Because of the potential for both HIV transmission and for serious adverse events in nursing infants with limited safety information in neonates, mothers should be counselled on the potential risks of breastfeeding if they are receiving Biktarvy.4.7 Effects on Ability to Drive and Use Machines
No studies on the effects of Biktarvy on the ability to drive and use machines have been performed.
4.8 Adverse Effects (Undesirable Effects)
As Biktarvy contains BIC, FTC, and TAF adverse reactions associated with these individual antiretroviral agents may be expected to occur with the fixed combination tablet.
Experience from clinical studies in treatment-naïve patients.
Assessment of adverse reactions is based on pooled data from two controlled clinical studies (Study 1489 and Study 1490) in which 1274 treatment-naïve patients received Biktarvy (N=634), abacavir (ABC)/DTG/lamivudine (3TC) (N=315) or dolutegravir (DTG) + FTC/TAF (N=325) in a double-blind phase through Week 144. After Week 144, patients in Studies 1489 and 1490 received open-label Biktarvy (N=1025) in an optional extension phase for an additional 96 weeks (end of study).
The most common adverse reactions (all Grades) and reported in ≥ 5% of patients in the Biktarvy group were diarrhoea and headache. The proportion of subjects who discontinued treatment with Biktarvy, ABC/DTG/3TC, or DTG + FTC/TAF due to adverse events, regardless of severity, was 0.9%, 1.6%, and 1.8%, respectively. Table 5 displays the frequency of adverse events (all Grades) greater than or equal to 3% in the Biktarvy group.
 Additional adverse reactions occurring in less than 3% of subjects administered Biktarvy in Studies 1489 and 1490 included abnormal dreams, vomiting, flatulence, dyspepsia, abdominal pain, and rash. The majority of adverse reactions occurred at severity Grade 1.
Additional adverse reactions occurring in less than 3% of subjects administered Biktarvy in Studies 1489 and 1490 included abnormal dreams, vomiting, flatulence, dyspepsia, abdominal pain, and rash. The majority of adverse reactions occurred at severity Grade 1.
No additional adverse reactions were identified in the open-label extension phases of Studies 1489 and 1490.
Laboratory abnormalities. The frequency of laboratory abnormalities (Grades 3-4) occurring in at least 2% of subjects receiving Biktarvy in Studies 1489 and 1490 are presented in Table 6.

Changes in serum creatinine.
BIC has been shown to increase serum creatinine due to inhibition of tubular secretion of creatinine without affecting renal glomerular function (see Section 5.2 Pharmacokinetic Properties). Increases in serum creatinine occurred by Week 4 of treatment and remained stable through Week 144. In Studies 1489 and 1490, median (Q1, Q3) serum creatinine increased by 0.11 (0.03, 0.19) mg/dL, 0.11 (0.04, 0.19) mg/dL, and 0.12 (0.06, 0.21) mg/dL from baseline to Week 144 in the Biktarvy, ABC/DTG/3TC, and DTG+FTC/TAF groups, respectively. There were no discontinuations due to renal adverse events through Week 144 in patients administered Biktarvy in clinical studies.
Changes in bilirubin.
In Studies 1489 and 1490, total bilirubin increases were observed in 17% of patients administered Biktarvy through Week 144. Increases were primarily Grade 1 (11%) and Grade 2 (4%) (≥ 1.0 to 2.5 x ULN), and were not associated with hepatic adverse reactions or other liver related laboratory abnormalities. Five patients administered Biktarvy (1%) had Grade 3 bilirubin increases that were not considered related to study drug. There were no discontinuations due to hepatic adverse events through Week 144 in Biktarvy clinical studies.
Experience from clinical studies in virologically suppressed patients.
No new adverse reactions to Biktarvy were identified through Week 48 in a controlled clinical study (GS-US-380-1844 ["Study 1844"]) of virologically suppressed patients who switched from regimens of DTG + ABC/3TC or ABC/DTG/3TC to Biktarvy (N=282).
No new adverse reactions to Biktarvy were identified through Week 48 in a controlled clinical study (GS-US-380-1878 ["Study 1878"]) of virologically suppressed patients who switched from regimens of ritonavir (RTV) or cobicistat (COBI) boosted atazanavir (ATV) or DRV, plus either FTC/TDF or ABC/3TC, to Biktarvy (N=290).
No new adverse reactions to Biktarvy were identified through Week 48 in a controlled clinical study (GS-US-380-4030 ["Study 4030"]) of virologically-suppressed patients who switched from DTG plus either FTC/TAF or FTC/TDF, to Biktarvy (N=284).
Experience from clinical studies in patients with renal impairment.
The safety of FTC + TAF (components of Biktarvy) was evaluated through 144 weeks in an open-label clinical study (Study 0112) in which 248 HIV-1 infected patients who were either treatment-naïve (N=6) or virologically suppressed (N=242) with mild to moderate renal impairment (eGFR by Cockcroft-Gault method 30-69 mL/min) received FTC+TAF in combination with EVG+COBI as a fixed-dose combination tablet. The safety profile of FTC+TAF in patients with mild to moderate renal impairment was similar to that from patients with normal renal function (see Section 5.1, Clinical trials).
Experience from clinical studies in paediatric patients.
The safety of Biktarvy was evaluated in 50 HIV-1 infected virologically suppressed patients between the ages of 12 to < 18 years (weighing ≥ 35 kg) through Week 48 and in 50 virologically suppressed patients between the ages of 6 to < 12 years (weighing ≥ 25 kg) through Week 24 in an open label clinical study (GS-US-380-1474 ["Study 1474"]). In this study, the safety profile of Biktarvy was similar to that in adults.
Experience from clinical studies in patients coinfected with HIV-1 and chronic hepatitis B.
The safety of TAF (a component of Biktarvy) for the treatment of chronic hepatitis B is based on data from two randomised, double-blind, active-controlled studies in adults with compensated liver disease (GS-US-320-0108 and GS-US-320-0110) (see Vemlidy (TAF) PI).
The safety of FTC + TAF in 72 HIV-suppressed adults coinfected with chronic hepatitis B was evaluated through Week 48 in an open-label clinical study (GS-US-292-1249) in which patients were switched from another antiretroviral regimen to FTC + TAF administered with elvitegravir (EVG) and cobicistat (COBI) as a fixed-dose combination tablet. The safety profile of this regimen in patients coinfected with HIV-1 and chronic hepatitis B was similar to that in patients with HIV-1 monoinfection (see Section 5.1, Clinical trials).
In 16 HIV/HBV coinfected adults administered Biktarvy (n = 8 HIV/HBV treatment naïve in Study 1490; n = 8 HIV/HBV suppressed in Study 1878), the adverse effects profile of Biktarvy was similar to that in patients with HIV-1 monoinfection. One patient in Study 1490 developed protocol-defined hepatic flare (ALT > 10 times upper limit of normal). ALT returned to normal limits without treatment interruption. Given limited data for use of Biktarvy in HBV coinfection, closely monitor coinfected patients (see Section 4.4 Special Warnings and Precautions for Use).
Reporting suspected adverse effects.
Reporting suspected adverse reactions after registration of the medicinal product is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions at http://www.tga.gov.au/reporting-problems.
Postmarketing experience.
In addition to adverse reactions from clinical studies, the following adverse reactions were identified during post-approval use with one or more of the components of Biktarvy. Because these reactions were reported voluntarily from a population of unknown size, estimates of frequency cannot be made.
Immune system disorders.
Autoimmune hepatitis (see Section 4.4 Special Warnings and Precautions for Use).
Skin and subcutaneous tissue disorders.
Angioedema, Stevens-Johnson syndrome, urticaria.
Psychological disorders.
Suicidal behaviour, anxiety, sleep disorders, depression.
Metabolic disorders.
Weight increased.
Renal and urinary disorders.
Acute renal failure, proximal renal tubulopathy, Fanconi syndrome.4.9 Overdose
If overdose occurs the patient must be monitored for evidence of toxicity. Treatment of overdose with Biktarvy consists of general supportive measures including monitoring of vital signs and ECG (QT interval) as well as observation of the clinical status of the patient.
For information on the management of overdose, contact the Poison Information Centre on 131126 (Australia) and 0800 764 766 (New Zealand).
Bictegravir.
Limited clinical experience is available at doses higher than the therapeutic dose of BIC. A single dose of 300 mg BIC (6 times the BIC dose in Biktarvy) was administered to 48 healthy subjects; no serious adverse reactions were reported. As BIC is highly bound to plasma proteins, it is unlikely that it will be significantly removed by haemodialysis or peritoneal dialysis.
Emtricitabine.
Limited clinical experience is available at doses higher than the therapeutic dose of FTC 200 mg. In one clinical pharmacology study single doses of FTC 1200 mg were administered to 11 patients. No severe adverse reactions were reported. The effects of higher doses are not known.
Haemodialysis treatment removes approximately 30% of the FTC dose over a 3-hour dialysis period starting within 1.5 hours of FTC dosing (blood flow rate of 400 mL/min and a dialysate flow rate of 600 mL/min). It is not known whether FTC can be removed by peritoneal dialysis.
Tenofovir alafenamide.
Limited clinical experience is available at doses higher than the therapeutic dose of TAF. A single supratherapeutic dose of 125 mg TAF was administered to 48 healthy patients, no serious adverse reactions were reported. The effects of higher doses are unknown. Tenofovir is efficiently removed by haemodialysis with an extraction coefficient of approximately 54%.5 Pharmacological Properties
5.1 Pharmacodynamic Properties
Pharmacotherapeutic group: Antivirals for treatment of HIV infections, combinations, ATC code: J05AR20.
Mechanism of action.
Bictegravir.
BIC is an integrase strand transfer inhibitor (INSTI) that binds to the integrase active site and blocks the strand transfer step of retroviral deoxyribonucleic acid (DNA) integration which is essential for the HIV replication cycle. BIC has activity that is specific to human immunodeficiency virus (HIV-1 and HIV-2).
Emtricitabine.
FTC is a nucleoside analogue of 2'-deoxycytidine. FTC is phosphorylated by cellular enzymes to form FTC triphosphate. FTC triphosphate inhibits HIV replication through incorporation into viral DNA by the HIV reverse transcriptase, which results in DNA chain-termination.
FTC has activity that is specific to human immunodeficiency virus (HIV-1 and HIV-2) and hepatitis B virus. FTC triphosphate is a weak inhibitor of mammalian DNA polymerases that include mitochondrial DNA polymerase γ and there was no evidence of toxicity to mitochondria in vitro and in vivo.
Tenofovir alafenamide.
TAF is a phosphonamidate prodrug of tenofovir (2'-deoxyadenosine monophosphate analogue). TAF is permeable into cells and due to increased plasma stability and intracellular activation through hydrolysis by cathepsin A, TAF is more efficient than tenofovir disoproxil fumarate (TDF) in loading tenofovir into peripheral blood mononuclear cells (PBMCs), including lymphocytes and macrophages. Intracellular tenofovir is subsequently phosphorylated to the pharmacologically active metabolite tenofovir diphosphate. Tenofovir diphosphate inhibits HIV replication through incorporation into viral DNA by the HIV reverse transcriptase, which results in DNA chain-termination.
Tenofovir has activity that is specific to human immunodeficiency virus (HIV-1 and HIV-2) and hepatitis B virus (HBV). In vitro studies have shown that both FTC and tenofovir can be fully phosphorylated when combined in cells. Tenofovir diphosphate is a weak inhibitor of mammalian DNA polymerases that include mitochondrial DNA polymerase γ and there is no evidence of mitochondrial toxicity in vitro based on several assays including mitochondrial DNA analyses.
Antiviral activity in vitro.
The triple combination of BIC, FTC, and TAF demonstrated synergistic antiviral activity in cell culture.
Bictegravir.
The antiviral activity of BIC against laboratory and clinical isolates of HIV-1 was assessed in lymphoblastoid cell lines, PBMCs, primary monocyte/macrophage cells, and CD4+ T-lymphocytes. The EC50 values for BIC for non-resistant strains were in the range of < 0.05 to 6.6 nanoM. The protein-adjusted EC95 of BIC was 361 nanoM (0.162 microgram/mL) for wild type HIV-1 virus.
BIC displayed antiviral activity in cell culture against HIV-1 groups (M, N, O), including subtypes A, B, C, D, E, F and G (EC50 values ranged from < 0.05 and 1.71 nanoM), and activity against HIV-2 (EC50 = 1.1 nanoM).
In a study of BIC with representatives from the major classes of approved anti-HIV agents (NRTIs [nucleoside reverse transcriptase inhibitors], NNRTIs [non-nucleoside reverse transcriptase inhibitors], INSTIs [integrase strand transfer inhibitors], and PIs [protease inhibitors]), additive to synergistic antiviral effects were observed. No antagonism was observed for these combinations.
Emtricitabine.
The in vitro antiviral activity of FTC against laboratory and clinical isolates of HIV was assessed in lymphoblastoid cell lines, the MAGI-CCR5 cell line, and PBMCs. The EC50 values for FTC were in the range of 0.0013 to 0.64 microM (0.0003 to 0.158 microgram/mL).
FTC displayed antiviral activity in vitro against HIV-1 clades A, C, D, E, F, and G (EC50 values ranged from 0.007 to 0.075 microM) and showed strain specific activity against HIV-2 (IC50 values ranged from 0.007 to 1.5 microM).
In two-drug combination studies of FTC with NRTIs, NNRTIs, and protease inhibitors (PI), and INSTIs, additive to synergistic effects were observed. No antagonism was observed for these combinations.
Tenofovir alafenamide.
The antiviral activity of TAF against laboratory and clinical isolates of HIV-1 subtype B was assessed in lymphoblastoid cell lines, PBMCs, primary monocyte/macrophage cells and CD4-T lymphocytes. The EC50 values for TAF were in the range of 2.0 to 14.7 nanoM.
TAF displayed antiviral activity in cell culture against all HIV-1 groups (M, N, O), including sub-types A, B, C, D, E, F, and G (EC50 values ranged from 0.10 to 12.0 nanoM) and strain specific activity against HIV-2 (EC50 values ranged from 0.91 to 2.63 nanoM).
In a study of TAF with a broad panel of representatives from the major classes of approved anti-HIV agents (NRTIs, NNRTIs, INSTIs, and PIs), additive to synergistic effects were observed. No antagonism was observed for these combinations.
Drug resistance.
In cell culture. Bictegravir.
HIV-1 isolates with reduced susceptibility to BIC have been selected in cell culture. In one selection, amino acid substitutions M50I and R263K emerged and phenotypic susceptibility to BIC was reduced 1.3-, 2.2-, and 2.9-fold for M50I, R263K, and M50I+R263K, respectively. In a second selection, amino acid substitutions T66I and S153F emerged and phenotypic susceptibility to BIC was shifted 0.4-, 1.9-, and 0.5-fold for T66I, S153F, and T66I+S153F, respectively.
Emtricitabine.
HIV-1 isolates with reduced susceptibility to FTC have been selected in cell culture. Genotypic analysis of these isolates showed that the reduced susceptibility to FTC was associated with a mutation in the HIV reverse transcriptase gene at codon 184 which resulted in an amino acid substitution of methionine by valine or isoleucine (M184V/I).
Tenofovir alafenamide.
HIV-1 isolates with reduced susceptibility to TAF have been selected in cell culture. HIV-1 isolates selected by TAF expressed a K65R mutation in HIV-1 RT; in addition, a K70E mutation in HIV-1 RT has been transiently observed. HIV-1 isolates with the K65R mutation have low-level reduced susceptibility to abacavir, FTC, tenofovir, and lamivudine. In vitro drug resistance selection studies with TAF have shown no development of high-level resistance after extended culture.
In clinical studies.
In treatment-naïve patients.
No patient receiving Biktarvy had HIV-1 with treatment emergent genotypic or phenotypic resistance to BIC, FTC, or TAF in the final resistance analysis population (n = 11 with data and HIV-1 RNA ≥ 200 copies/mL at the time of confirmed virologic failure or early study drug discontinuation) in a pooled analysis of antiretroviral-naïve patients through Week 144 of the double-blind phase (N=634) or 96 weeks of the extension phase (N=1065) of Studies 1489 and 1490.
In virologically suppressed patients.
No patient receiving Biktarvy had HIV-1 with treatment emergent genotypic or phenotypic resistance to BIC, FTC, or TAF in the final resistance analysis population (n = 2 with HIV-1 RNA ≥ 200 copies/mL at the time of confirmed virologic failure, Week 48, or early study drug discontinuation) of 282 virologically-suppressed patients who switched from DTG + ABC/3TC or ABC/DTG/3TC to Biktarvy (Study 1844).
No patient receiving Biktarvy had HIV-1 with treatment emergent genotypic or phenotypic resistance to BIC, FTC, or TAF in the resistance analysis population (n = 1 with HIV-1 RNA ≥ 200 copies/mL at the time of confirmed virologic failure, Week 48, or early study drug discontinuation) of 290 virologically-suppressed patients who switched from regimens of RTV or COBI boosted ATV or DRV, plus either FTC/TDF or ABC/3TC, to Biktarvy (Study 1878).
No patient receiving Biktarvy had HIV-1 with treatment-emergent genotypic or phenotypic resistance to BIC, FTC, or TAF through the end of the blinded treatment phase of 284 virologically-suppressed patients who switched from DTG plus either FTC/TAF or FTC/TDF, to Biktarvy (Study 4030).
In patients coinfected with HIV-1 and chronic hepatitis B.
In a clinical study of patients coinfected with HIV-1 and chronic hepatitis B who received FTC + TAF in combination with EVG+COBI as a fixed-dose combination tablet for 48 weeks (GS-US-292-1249, N = 72), no subject had HIV or HBV emergent resistance to FTC, TAF, or EVG.
Cross-resistance.
Bictegravir. Integrase strand transfer inhibitor-resistant mutant HIV-1 strains.
The susceptibility of BIC was tested against 64 INSTI-resistant clinical isolates (20 with single substitutions and 44 with 2 or more substitutions). Fifty of the 64 INSTI-resistant clinical isolates had ≤ 2.5-fold phenotypic change to BIC and were assessed as sensitive. All single and double mutants of these isolates lacking Q148H/K/R, and 10 of 24 Q148H/K/R containing isolates with or without additional INSTI resistance associated substitutions also had ≤ 2.5-fold reduced susceptibility to BIC. Reduced susceptibility to BIC of > 2.5 fold was found for 14 of the 24 Q148H/R/K containing isolates that also had G140A/C/S substitutions in integrase; 9 of those 14 isolates had additional mutations at L74M, T97A, or E138A/K. In addition, site-directed mutants with G118R and T97A+G118R had 3.4- and 2.8-fold reduced susceptibility to BIC, respectively. A clinical isolate carrying the T66I+E138K+Q148K triple mutation conferred substantial resistance to BIC (44-fold) in vitro.
Reverse transcriptase inhibitor- and protease inhibitor-resistant strains.
BIC demonstrated equivalent antiviral activity against 5 NNRTI-resistant, 3 NRTI-resistant, and 4 PI-resistant HIV-1 mutant clones compared with the wild-type strain.
Emtricitabine. FTC-resistant isolates (M184V/I) were cross-resistant to 3TC but retained sensitivity to didanosine, d4T, tenofovir and AZT.
Viruses harbouring mutations conferring reduced susceptibility to d4T and AZT-thymidine analogue-associated mutations - TAMs (M41L, D67N, K70R, L210W, T215Y/F, K219Q/E) or didanosine (L74V) remained sensitive to FTC. HIV-1 containing the K103N mutation or substitutions associated with resistance to NNRTI were susceptible to FTC.
Tenofovir alafenamide. The K65R and K70E mutations result in reduced susceptibility to abacavir, didanosine, lamivudine, FTC, and tenofovir, but retain sensitivity to zidovudine.
Multinucleoside resistant HIV-1 with a T69S double insertion mutation or with a Q151M mutation complex including K65R showed reduced susceptibility to TAF.
HIV-1 containing the K103N or Y181C mutations associated with resistance to NNRTIs were susceptible to TAF.
HIV-1 containing mutations associated with resistance to PIs, such as M46I, I54V, V82F/T, and L90M were susceptible to TAF.
Effects on electrocardiogram.
Bictegravir.
In a thorough QT/QTc study in 48 healthy subjects, BIC at supratherapeutic doses of 1.5 and 6 times the recommended therapeutic dose did not affect the QT/QTc interval and did not prolong the PR interval.
Tenofovir alafenamide.
In a thorough QT/QTc study in 48 healthy subjects, TAF at the therapeutic dose or at a supratherapeutic dose approximately 5 times the recommended therapeutic dose did not affect the QT/QTc interval and did not prolong the PR interval. The effect of the other component, FTC, or the combination of FTC and TAF on the QT interval is not known.
Effects on serum creatinine.
The effect of BIC on renal function was evaluated in a randomised, blinded, parallel, placebo-controlled trial in 40 healthy subjects who received BIC 75 mg (n = 20) or placebo (n = 20) once daily with food for 14 days. Mean change from baseline in serum creatinine in the BIC group was 0.1 mg/dL on Days 7 and 14. BIC did not have a clinically significant effect on the estimated glomerular filtration rate or on the actual glomerular filtration rate (determined by the clearance of probe drug, iohexol) compared with placebo.
Clinical trials.
The efficacy and safety of Biktarvy in HIV-1 infected, treatment-naïve adults are based on 48-week and 144-week data from two randomised, double-blind, active-controlled studies, GS-US-380-1489 ("Study 1489") (N=629) and GS-US-380-1490 ("Study 1490") (N=645). The safety of Biktarvy is also supported by data from patients who received open-label Biktarvy (N=1025) after Week 144 in an extension phase of Studies 1489 and 1490 for an additional 96 weeks (end of study).
The efficacy and safety of Biktarvy in virologically-suppressed HIV-1 infected adults are based on 48-week data from a randomised, double-blind, active-controlled study, GS-US-380-1844 ("Study 1844") (N = 563); and a randomised, open label, active-controlled study, GS-US-380-1878 ("Study 1878") (N=577), and a randomised, double-blind, active-controlled study, GS-US-380-4030 ("Study 4030") (N = 284; 47 of whom had the FTC-associated resistance mutation M184V/I in HIV at baseline).
The efficacy and safety of FTC + TAF (components of Biktarvy) in HIV-1 infected, virologically-suppressed patients with mild to moderate renal impairment is based on 144-week data from an open-label study, Study 0112 (N=242). The efficacy and safety of FTC+TAF (components of Biktarvy) in HIV-1 infected, virologically-suppressed patients with ESRD on chronic haemodialysis is based on data from a single arm, open-label study, GS-US-292-1825 ("Study 1825") in which 55 patients were administered FTC+TAF in combination with EVG+COBI through Week 96, followed by an extension phase in which 10 patients were administered Biktarvy for 48 weeks.
The efficacy and safety of Biktarvy in HIV-1 infected paediatric patients is based on data from open-label Study 1474, in virologically-suppressed paediatric patients between the ages of 12 to < 18 years (≥ 35 kg) (N=50) and between the ages of 6 to < 12 years (≥ 25 kg) (N=50).
The efficacy and safety of FTC+TAF (components of Biktarvy) in adult patients coinfected with HIV-1 and chronic hepatitis B are based on 48-week data from an open-label study, GS-US-292-1249 ("Study 1249") (N=72). The efficacy and safety of Biktarvy in adult patients coinfected with HIV-1 and chronic hepatitis B are also supported by 48 and 144-week data in 8 HIV/HBV coinfected adults treated with Biktarvy in Study 1490 and 48-week data in 8 HIV/HBV coinfected adults treated with Biktarvy in Study 1878.
Treatment-naïve patients.
Study 1489 and 1490. In Study 1489, patients were randomised in a 1:1 ratio to receive either Biktarvy (N=314) or ABC/DTG/3TC (600/50/300 mg) (N=315) once daily. In Study 1490, patients were randomised in a 1:1 ratio to receive either Biktarvy (N=320) or DTG + FTC/TAF (50+200/25 mg) (N=325) once daily.
In Studies 1489 and 1490, the mean age was 35 years (range 18-77), 89% were male, 58% were White, 33% were Black, and 3% were Asian. 24% percent of patients identified as Hispanic/Latino. The mean baseline plasma HIV-1 RNA was 4.4 log10 copies/mL (range 1.3-6.6). The mean baseline CD4+ cell count was 460 cells/mm3 (range 0-1636) and 11% had CD4+ cell counts less than 200 cells/mm3. 18% of patients had baseline viral loads greater than 100,000 copies/mL. In Study 1490, 14 patients had HIV/HBV coinfection and 10 patients had HIV/HCV coinfection at baseline. In Study 1489, 4 patients had HIV/HCV coinfection at baseline. In both studies, patient randomisation were stratified by baseline HIV-1 RNA (less than or equal to 100,000 copies/mL, greater than 100,000 copies/mL to less than or equal to 400,000 copies/mL, or greater than 400,000 copies/mL), by CD4 count (less than 50 cells/microL, 50-199 cells/microL, or greater than or equal to 200 cells/microL), and by region (US or ex-US).
Treatment outcomes of Studies 1489 and 1490 through Weeks 48 and 144 are presented in Table 7.
 Biktarvy was noninferior in achieving HIV-1 RNA < 50 copies/mL at both Week 48 and 144 when compared to ABC/DTG/3TC and to DTG+FTC/TAF, respectively. Treatment outcomes were similar among treatment groups across subgroups by age, sex, race, baseline viral load, and baseline CD4+ cell count.
Biktarvy was noninferior in achieving HIV-1 RNA < 50 copies/mL at both Week 48 and 144 when compared to ABC/DTG/3TC and to DTG+FTC/TAF, respectively. Treatment outcomes were similar among treatment groups across subgroups by age, sex, race, baseline viral load, and baseline CD4+ cell count.
In Studies 1489 and 1490, the mean increase from baseline in CD4+ count at Week 144 was 288, 317, and 289 cells/mm3 in the pooled Biktarvy, ABC/DTG/3TC, and DTG+FTC/TAF groups, respectively.
Bone mineral density.
In Study 1489, bone mineral density (BMD) change from baseline to Week 144 was assessed by dual-energy X-ray absorptiometry (DXA). In patients who had both baseline and Week 144 hip and lumbar spine BMD measurements (N= 236 and 243 in the Biktarvy group and N= 240 and 244 in the ABC/DTG/3TC group, for hip and lumbar spine, respectively), mean percentage changes in BMD were similar in the Biktarvy group compared to the ABC/DTG/3TC group for hip (-1.0% vs. -1.3%) and lumbar spine (-0.4% vs. 0.04%).
Patients coinfected with HIV-1 and chronic hepatitis B.
In Study 1490, 7 of 8 patients with HIV/HBV coinfection at baseline who were randomised to receive Biktarvy were HBV suppressed (HBV DNA < 29 IU/mL) and had HIV-1 RNA < 50 copies/mL at Week 48. One patient had missing HBV DNA data at Week 48. At Week 144, 5 patients were HBV suppressed and had HIV-1 RNA < 50 copies/mL. Three patients had missing HBV DNA data at Week 144 (1 lost to follow-up prior to Week 48, 1 lost to follow-up after Week 72, and 1 lost to follow-up after week 120).
Virologically-suppressed patients.
Study 1844 and study 1878. In Study 1844, the efficacy and safety of switching from a regimen of DTG + ABC/3TC or ABC/DTG/3TC to Biktarvy were evaluated in a randomised, double-blind study of virologically-suppressed (HIV-1 RNA < 50 copies/mL) HIV-1 infected adults (N=563). Patients must have been stably suppressed (HIV-1 RNA < 50 copies/mL) on their baseline regimen for at least 3 months prior to study entry. Patients were randomised in a 1:1 ratio to either switch to Biktarvy at baseline (N=282), or stay on their baseline antiretroviral regimen (N=281). Patients had a mean age of 45 years (range 20-71), 89% were male, 73% were White, and 22% were Black. 17% of patients identified as Hispanic/Latino. The mean baseline CD4+ cell count was 723 cells/mm3 (range 124-2444).
In Study 1878, the efficacy and safety of switching from either ABC/3TC or FTC/TDF (200/300 mg) plus ATV or DRV (boosted by either COBI or RTV) to Biktarvy were evaluated in a randomised, open-label study of virologically-suppressed HIV-1 infected adults (N=577). Patients must have been stably suppressed on their baseline regimen for at least 6 months and must not have been previously treated with any INSTI. Patients were randomised in a 1:1 ratio to either switch to Biktarvy (N=290), or stay on their baseline antiretroviral regimen (N=287). Patients had a mean age of 46 years (range 20-79), 83% were male, 66% were White, and 26% were Black. 19% of patients identified as Hispanic/Latino. The mean baseline CD4+ cell count was 663 cells/mm3 (range 62-2582). Patients were stratified by prior treatment regimen. At screening, 15% of patients were receiving ABC/3TC plus ATV or DRV (boosted by either COBI or RTV) and 85% of patients were receiving FTC/TDF plus ATV or DRV (boosted by either COBI or RTV).
Treatment outcomes of Studies 1844 and 1878 through Week 48 are presented in Table 8.
 In Study 1844, at Week 48, switching to Biktarvy was noninferior to remaining on ABC/DTG/3TC. The percentages of patients with HIV-1 RNA ≥ 50 copies/mL and who maintained HIV-1 RNA < 50 copies/mL were similar between the Biktarvy and ABC/DTG/3TC groups. Treatment outcomes between treatment groups were similar across subgroups by age, sex and race. The mean change from baseline in CD4+ count at Week 48 was -31 cells/mm3 in patients who switched to Biktarvy and 4 cells/mm3 in patients who stayed on ABC/DTG/3TC.
In Study 1844, at Week 48, switching to Biktarvy was noninferior to remaining on ABC/DTG/3TC. The percentages of patients with HIV-1 RNA ≥ 50 copies/mL and who maintained HIV-1 RNA < 50 copies/mL were similar between the Biktarvy and ABC/DTG/3TC groups. Treatment outcomes between treatment groups were similar across subgroups by age, sex and race. The mean change from baseline in CD4+ count at Week 48 was -31 cells/mm3 in patients who switched to Biktarvy and 4 cells/mm3 in patients who stayed on ABC/DTG/3TC.
In Study 1878, at Week 48, switching to Biktarvy was noninferior to remaining on an ATV- or DRV-based regimen. The percentages of patients with HIV-1 RNA ≥ 50 copies/mL and who maintained HIV-1 RNA < 50 copies/mL were similar between the Biktarvy and ATV- or DRV-based regimen groups. Treatment outcomes between treatment groups were similar across subgroups by age, sex and race. The mean change from baseline in CD4+ count at Week 48 was 25 cells/mm3 in patients who switched to Biktarvy and 0 cells/mm3 in patients who stayed on their baseline regimen.
In Study 4030, the efficacy and safety of switching from DTG plus either FTC/TAF or FTC/TDF, to Biktarvy were evaluated in a randomised, double-blind study of virologically suppressed HIV-1 infected adults. Patients must have been stably suppressed (HIV-1 RNA less than 50 copies per mL) on their baseline regimen for at least 6 months (if documented or suspected nucleoside reverse transcriptase inhibitor [NRTI] resistance), or at least 3 months (if no documented or suspected NRTI resistance) prior to study entry. Patients were randomised to switch to Biktarvy (N=284) or to continue on DTG plus FTC/TAF (N=281). The primary endpoint was the proportion of patients with HIV RNA ≥ 50 copies/mL at Week 48. At Week 48, Biktarvy was noninferior to DTG+FTC/TAF; the proportion of patients with HIV-1 RNA ≥ 50 copies/mL was 0.4% (1/284) in the Biktarvy group and 1.1% (3/281) in the DTG+FTC/TAF group (difference: -0.7% [95% CI: -2.8%, 1.0%]).
Of the patients receiving Biktarvy, 47 had HIV-1 with the FTC-associated M184V/I resistance mutation at baseline. At Week 48, no patient with M184V/I had HIV RNA ≥ 50 copies/mL. Eighty-nine percent (42/47) of patients with M184V/I remained suppressed (HIV-1 RNA < 50 copies/mL) and 11% (5/47 patients) did not have virologic data at Week 48 due to discontinuation.
Bone mineral density.
In Study 1844, BMD change from baseline to Week 48 was assessed by DXA. In patients who had both baseline and Week 48 hip and lumbar spine BMD measurements (N=229 and 233 in the Biktarvy group and N=242 and 244 in the ABC/DTG/3TC group, for hip and lumbar spine, respectively), mean percentage increases in BMD were similar in the Biktarvy group compared to the ABC/DTG/3TC group for hip (0.2% vs. 0.3%) and lumbar spine (0.7% vs. 0.4%).
Patients coinfected with HIV-1 and chronic hepatitis B.
In Study 1878, at Week 48, 100% (8/8) of the patients coinfected with HIV/HBV at baseline in the Biktarvy maintained HBV DNA < 29 IU/mL (missing = excluded analysis) and HIV RNA < 50 copies/mL.
Paediatric patients.
In Study 1474, the efficacy, safety, and pharmacokinetics of Biktarvy in HIV-1 infected virologically-suppressed paediatric patients between the ages of 12 to < 18 years (≥ 35 kg) (N=50) and between the ages of 6 to < 12 years (≥ 25 kg) (N=50) were evaluated.
Cohort 1: virologically suppressed adolescents (12 to < 18 years; ≥ 35 kg).
Patients in cohort 1 had a mean age of 14 years (range: 12 to 17) and a mean baseline weight of 51.7 kg (range: 35 to 123), 64% were female, 27% were Asian, and 65% were black. At baseline, median CD4+ cell count was 750 cells/mm3 (range: 337 to 1207), and median CD4+% was 33% (range: 19% to 45%).
After switching to Biktarvy, 98% (49/50) of patients in cohort 1 remained suppressed (HIV-1 RNA < 50 copies/mL) at Week 48. The mean change from baseline in CD4+ cell count at Week 48 was -22 cells/mm3. Two of 50 subjects met the criteria for inclusion in the resistance analysis population through Week 48. No emergent resistance to Biktarvy was detected through Week 48.
Cohort 2: virologically suppressed children (6 to < 12 years; ≥ 25 kg).
Patients in cohort 2 had a mean age of 10 years (range: 6 to 11) and a mean baseline weight of 31.9 kg (range: 25 to 69), 54% were female, 22% were Asian and 72% were black. At baseline, median CD4+ cell count was 898 cells/mm3 (range 390 to 1991) and median CD4+% was 37% (range: 19% to 53%).
After switching to Biktarvy, 100% (50/50) of patients in cohort 2 remained suppressed (HIV-1 RNA < 50 copies/mL) at Week 24. The mean change from baseline in CD4+ cell count at Week 24 was -24 cells/mm3. No patient qualified for resistance analysis through Week 24.
5.2 Pharmacokinetic Properties
The pharmacokinetic (PK) properties of Biktarvy components are provided in Table 9.
 The multiple dose pharmacokinetic parameters of the components of Biktarvy are provided in Table 10.
The multiple dose pharmacokinetic parameters of the components of Biktarvy are provided in Table 10.

Pharmacokinetics in special populations.
Age, gender and ethnicity. Population analyses using pooled pharmacokinetic data from adult trials did not identify any clinically relevant differences due to age, gender or race on the exposures of BIC, FTC, or TAF.
Mean BIC Ctrough was lower in 50 paediatric patients aged 12 to < 18 years (≥ 35 kg) who received Biktarvy in Study 1474 relative to adults following administration of Biktarvy, but was not considered clinically significant based on exposure-response relationships; exposures of FTC and TAF in these paediatric patients were similar to those in adults. Mean BIC Cmax, and exposures of FTC and TAF (AUC and Cmax), achieved in 50 paediatric patients between the ages of 6 to < 12 years (≥ 25 kg) who received Biktarvy in Study 1474 were higher than exposures in adults; however, the increase was not considered clinically significant as the safety profiles were similar in adult and paediatric patients.
Patients with impaired renal function. No clinically relevant differences in BIC, TAF, or tenofovir pharmacokinetics were observed between healthy subjects and subjects with severe renal impairment (estimated creatinine clearance ≥ 15 and < 30 mL /minute) in Phase 1 studies. In a separate Phase 1 study of FTC alone, FTC exposures were increased in subjects with severe renal impairment. The safety of Biktarvy has not been established in subjects with estimated creatinine clearance ≥ 15 mL and < 30 mL/min.
Exposures of FTC and tenofovir in 12 patients with ESRD (estimated creatinine clearance < 15 mL/min) on chronic haemodialysis who received FTC+TAF in combination with EVG+COBI as a fixed-dose combination tablet in Study 1825 were significantly higher than in patients with normal renal function. However, the safety profile of FTC+TAF in patients with ESRD on chronic haemodialysis who received FTC+TAF in combination with EVG+COBI was similar to that in patients with normal renal function. No clinically relevant differences in TAF pharmacokinetics were observed in patients with ESRD as compared to those with normal renal function.
In the extension phase of Study 1825, a lower BIC Ctrough was observed in patients with ESRD who received Biktarvy compared to patients with normal renal function, but this difference was not considered clinically relevant.
There are no pharmacokinetic data on BIC, FTC or TAF in patients with creatinine clearance < 15 mL/min not on chronic haemodialysis.
Patients with hepatic impairment. Bictegravir.
Clinically relevant changes in the pharmacokinetics of BIC were not observed in subjects with moderate hepatic impairment.
Emtricitabine.
The pharmacokinetics of FTC has not been studied in subjects with hepatic impairment; however, FTC is not significantly metabolised by liver enzymes, so the impact of liver impairment should be limited.
Tenofovir alafenamide.
Clinically relevant changes in the pharmacokinetics of TAF or its metabolite tenofovir were not observed in patients with mild, moderate, or severe hepatic impairment; no TAF dose adjustment is required in patients with hepatic impairment.
Hepatitis B and/or hepatitis C virus coinfection. Pharmacokinetics of BIC, FTC and TAF have not been fully evaluated in hepatitis B and/or C coinfected patients.
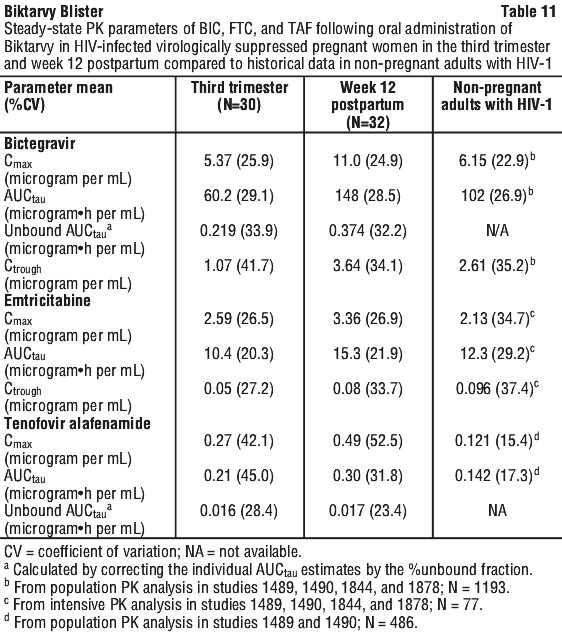
Pregnancy. Plasma exposures of BIC, FTC, and TAF were lower during pregnancy as compared to postpartum, whereas exposures during postpartum were generally higher than in non-pregnant adults (Table 11). Exposures were generally similar between the second and third trimesters of pregnancy; exposures were also generally similar between Weeks 6 and 12 postpartum. Based on exposure-response relationships for BIC, FTC, and TAF, the exposure changes during pregnancy are not considered to be clinically relevant (Table 11); however, specific dosage adjustments for coadministered oral medications or supplements containing polyvalent cations are recommended in pregnant patients (see Section 4.5; Section 4.6).
5.3 Preclinical Safety Data
Genotoxicity.
No genotoxicity studies have been conducted with BIC, FTC and TAF in combination.
Bictegravir.
BIC was not mutagenic in bacteria or clastogenic in human lymphocytes in vitro and in a rat micronucleus test in vivo.
Emtricitabine.
FTC was not mutagenic in bacteria or mouse lymphoma cell assays in vitro nor clastogenic in the mouse micronucleus test in vivo.
Tenofovir alafenamide.
TAF was not genotoxic in the reverse mutation bacterial test (Ames test), mouse lymphoma or rat micronucleus assays.
Carcinogenicity.
No carcinogenicity studies have been conducted with BIC, FTC and TAF in combination.
Bictegravir.
BIC was not carcinogenic in a 6-month rasH2 transgenic mouse study at doses of up to 100 mg/kg/day in males and 300 mg/kg/day in females, respectively. This resulted in exposures of approximately 15 (males) and 23 (females) times the exposure in humans at the recommended dose. BIC was also not carcinogenic in a 104 week rat study at doses up to 300 mg/kg/day. This resulted in an exposure of approximately 31 times the exposure in humans at the recommended dose.
Emtricitabine.
In long-term oral carcinogenicity studies conducted with FTC, no drug-related increases in tumour incidence were found in mice at doses up to 750 mg/kg/day (32 times the human systemic exposure (AUC) at the therapeutic dose of 200 mg/day) or in rats at doses up to 600 mg/kg/day (38 times the human systemic exposure at the therapeutic dose).
Tenofovir alafenamide.
Because there is a lower tenofovir exposure in rats and mice after TAF administration compared to TDF, carcinogenicity studies were conducted only with TDF. Long-term oral carcinogenicity studies of TDF in mice and rats were carried out at exposures up to approximately 10 times (mice) and 4 times (rats) those observed in humans at the 300 mg therapeutic dose of TDF for HIV-1 infection. At the high dose in female mice, liver adenomas were increased at tenofovir exposures 10 times (300 mg TDF) and 151 times (Biktarvy) the exposure observed in humans. In rats, the study was negative for carcinogenic findings.6 Pharmaceutical Particulars
6.1 List of Excipients
Croscarmellose sodium, magnesium stearate, and microcrystalline cellulose.
The tablets are film-coated with a coating material containing iron oxide black, iron oxide red, macrogol 3350, polyvinyl alcohol, purified talc, and titanium dioxide.
See Section 2 Qualitative and Quantitative Composition.
6.2 Incompatibilities
Not applicable.
6.3 Shelf Life
In Australia, information on the shelf life can be found on the public summary of the Australian Register of Therapeutic Goods (ARTG). The expiration date can be found on the packaging.
6.4 Special Precautions for Storage
Biktarvy should be stored below 25°C.
6.5 Nature and Contents of Container
Biktarvy tablets are supplied in blister packs consisting of polyvinyl chloride/polyethylene/polychlorotrifluoroethylene (PVC/PE/PCTFE) film, sealed to aluminium foil lidding material fitted with a molecular sieve desiccant within each blister cavity. Each carton contains 30 tablets (4 x blister strips containing 7 tablets and 1 x blister strip containing 2 tablets).
6.6 Special Precautions for Disposal
In Australia, any unused medicine or waste material should be disposed of by taking to your local pharmacy.
6.7 Physicochemical Properties
Chemical structure.
Bictegravir.
The chemical name of bictegravir sodium is 2,5-Methanopyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazepine-10-carboxamide, 2,3,4,5,7,9,13,13a-octahydro-8-hydroxy-7,9-dioxo-N-[(2,4,6-trifluorophenyl)methyl]-, sodium salt (1:1), (2R,5S,13aR)-.
Bictegravir sodium has a molecular formula of C21H17F3N3NaO5 and a molecular weight of 471.4 and has the following structural formula:
 Bictegravir sodium is a white to off-white to yellow solid with a solubility of 0.1 mg/mL in water at 20°C.
Bictegravir sodium is a white to off-white to yellow solid with a solubility of 0.1 mg/mL in water at 20°C.
Emtricitabine.
FTC is a synthetic nucleoside analog of cytidine. The chemical name of FTC is 5-fluoro-1-(2R,5S)-[2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine. FTC is the (-) enantiomer of a thio analog of cytidine, which differs from other cytidine analogs in that it has a fluorine in the 5-position.
It has a molecular formula of C8H10FN3O3S and a molecular weight of 247.2. It has the following structural formula:
 FTC is a white to off-white crystalline powder with a solubility of approximately 112 mg/mL in water at 25°C. The partition coefficient (log p) for FTC is -0.43 and the pKa is 2.65.
FTC is a white to off-white crystalline powder with a solubility of approximately 112 mg/mL in water at 25°C. The partition coefficient (log p) for FTC is -0.43 and the pKa is 2.65.
Tenofovir alafenamide fumarate.
TAF is converted in vivo to tenofovir, an acyclic nucleoside phosphonate (nucleotide) analog of adenosine 5'-monophosphate. The chemical name of tenofovir alafenamide fumarate is L-Alanine, N-[(S)-[[(1R)-2-(6-amino-9H-purin-9-yl)-1-methylethoxy]methyl]phenoxyphosphinyl]-, 1-methylethyl ester, (2E)-2-butenedioate (2:1).
It has an empirical formula of C21H29O5N6P.½(C4H4O4) and a molecular weight of 534.5. It has the following structural formula:
 TAF is a white to off-white or tan powder with a solubility of 4.7 mg/mL in water at 20°C.
TAF is a white to off-white or tan powder with a solubility of 4.7 mg/mL in water at 20°C.
CAS number.
Bictegravir sodium.
CAS number: 1807988-02-8.
Emtricitabine.
CAS number: 143491-57-0.
Tenofovir alafenamide.
CAS number: 379270-37-8.
Tenofovir alafenamide fumarate.
CAS number: 1392275-56-7.7 Medicine Schedule (Poisons Standard)
Schedule 4 - Prescription Only Medicine.
Summary Table of Changes

