
What is in this leaflet
Read all of this leaflet carefully before you start taking this medicine. This leaflet answers some of the common questions about BIKTARVY tablets. It does not contain all of the available information.
It does not take the place of talking to your doctor or pharmacist about your medical condition or treatment. If you have further questions, please ask your doctor or your pharmacist.
This medicine has been prescribed for you personally and you should not pass it on to others. It may harm them, even if their symptoms are the same as yours.
Medicines are sometimes prescribed for conditions that are not mentioned in this leaflet.
Keep this leaflet with your BIKTARVY medicine. You may need to read it again.
What is BIKTARVY
How BIKTARVY works
BIKTARVY tablets consist of the following medicines:
- bictegravir
- emtricitabine
- tenofovir alafenamide
Bictegravir belongs to a group of antiretroviral medicines known as integrase strand transfer inhibitors (INSTI).
Emtricitabine and tenofovir alafenamide belong to a group of antiviral medicines known as nucleoside and nucleotide reverse transcriptase inhibitors (NRTI) and (NtRTI) respectively.
These are combined in one tablet to help control Human Immunodeficiency Virus 1 (HIV-1) infection in adults and children weighing at least 25 kg.
When used to treat HIV-1 infection
BIKTARVY lowers the amount of HIV in the blood (viral load). BIKTARVY may also help to increase the number of T cells (CD4+ cells), allowing your immune system to improve. Lowering the amount of HIV in the blood lowers the chance of death or infections that happen when your immune system is weak (opportunistic infections).
HIV infection destroys CD4+ T cells, which are important to the immune system. The immune system helps fight infection. After a large number of T cells are destroyed, acquired immune deficiency syndrome (AIDS) may develop.
BIKTARVY is for people who do not have a HIV virus resistant to BIKTARVY.
Emtricitabine and tenofovir alafenamide are also components of DESCOVY, GENVOYA, ODEFSEY tablets. VEMLIDY tablets contain tenofovir alafenamide.
Use in children
BIKTARVY is used to treat HIV-1 infection in adults and children weighing at least 25 kg.
The use of BIKTARVY in children and adolescents under 18 years of age has not yet been established.
Does BIKTARVY cure HIV, AIDS or HBV
BIKTARVY does not cure HIV infection, AIDS or HBV infection.
The long-term effects of BIKTARVY are not known at this time.
People taking BIKTARVY or any other medication for HIV may still get opportunistic infections or other conditions that happen with HIV infection.
Opportunistic infections are infections that develop because the immune system is weakened. Some of these conditions are pneumonia, herpes virus infections, and Mycobacterium avium complex (MAC) infection.
This medicine is only available from a pharmacist after it has been prescribed by a doctor who specialises in the treatment of HIV infection.
If you wish to continue receiving treatment with BIKTARVY it is important you remain under the care of a hospital or doctor who specialises in the treatment of HIV infection.
Does BIKTARVY reduce the risk of passing HIV to others
Effective viral suppression with BIKTARVY will substantially reduce the risk of passing HIV to others. However, a residual risk cannot be excluded.
Discuss with your doctor the precautions needed to avoid infecting other people.
For your health and the health of others, it is important to always practice safer sex by using a latex or polyurethane condom or other barrier to lower the chance of sexual contact with semen, vaginal secretions, or blood.
Never re-use or share needles.
Before you take BIKTARVY
When you must not take it
Together with your doctor, you need to decide whether BIKTARVY is right for you.
Do not take BIKTARVY if you are allergic to:
- bictegravir
- emtricitabine
- tenofovir
- or any of the other ingredients of BIKTARVY.
The ingredients of BIKTARVY are listed in the product description section of this leaflet.
Do not take BIKTARVY if you are already taking any other medicines that contain the same active ingredients.
Do not take BIKTARVY if you are taking other medicines that contain:
- tenofovir disoproxil fumarate (e.g. Truvada, Viread)
- tenofovir alafenamide (e.g. Descovy, Genvoya, Odefsey, Vemlidy)
- emtricitabine (e.g. Descovy, Emtriva, Genvoya, Odefsey)
- lamivudine (e.g. Combivir, Triumeq)
Do not take BIKTARVY to treat your HIV infection if you are also taking dofetilide to treat heart conditions.
Do not take BIKTARVY to treat your HIV infection if you are also taking rifampicin to treat infections.
Do not take BIKTARVY to treat your HIV infection if you are also taking adefovir dipivoxil to treat your hepatitis B virus (HBV) infection.
Before you start to take it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have, or have had, any of the following medical conditions:
- kidney problems or are undergoing kidney dialysis treatment.
- liver problems, including hepatitis B or C virus infection.
Tell your doctor if you are pregnant, or likely to become pregnant during your course of medication. We do not know if BIKTARVY can harm your unborn child. You and your doctor will need to decide if BIKTARVY is right for you.
Tell your doctor if you are breastfeeding, or likely to breastfeed during your course of medication. You should not breastfeed if you are HIV-positive because of the chance of passing the HIV virus to your baby. At least one of the active substances in this medicine (emtricitabine) has been found in breast milk at low concentrations. Talk with your doctor about the best way to feed your baby.
Taking other medicines
Tell your doctor or pharmacist if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop.
Some medicines and BIKTARVY may interfere with each other. These include:
- carbamazepine (e.g. Tegretol)
- oxcarbazepine (e.g. Trileptal)
- phenobarbital or phenytoin (e.g. Dilantin)
- rifabutin (e.g. Mycobutin)
- rifapentine (e.g. Priftin)
- boceprevir (e.g. Victrelis)
- St John’s Wort or products containing St John’s Wort
- atazanavir (e.g. Reyataz)
This is not a complete list of medicines that you should tell your doctor about.
These medicines may be affected by BIKTARVY or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
For this reason, it is very important to let your doctor or pharmacist know what medications, herbal supplements, or vitamins you are taking.
Know the medicines you take. Keep a list of medicines and show it to your doctor and pharmacist when you get a new medicine.
Do not start any new medicines while you are taking BIKTARVY without first talking with your doctor or pharmacist.
How to take BIKTARVY
Take the exact amount of BIKTARVY your doctor has prescribed for you.
Never change the dose on your own.
Do not stop this medicine unless your healthcare provider tells you to stop.
How much to take
The usual dose is one BIKTARVY tablet orally, once daily.
Take BIKTARVY with or without food.
Do not chew, crush or split the tablet.
When to take it
If you are taking an antacid (such as aluminium/magnesium hydroxide), a mineral supplement or vitamin (containing calcium or iron), ulcer-healing medication (such as sucralfate), or a buffered medication (containing calcium carbonate), take BIKTARVY at least 2 hours before taking these medications.
Alternatively, you can take the medication and BIKTARVY together with food.
If you forget to take it
Do not miss a dose of BIKTARVY.
If you forget to take BIKTARVY, take your missed dose right away unless it is almost time for your next dose.
Do not take a double dose to make up for a forgotten dose. Continue with your regular dosing schedule.
When your BIKTARVY supply starts to run low, get more from your doctor or pharmacy. This is very important because the amount of virus in your blood may increase if the medicine is stopped for even a short time. The virus may develop resistance to BIKTARVY and become harder to treat.
If you take too much (overdose)
Immediately telephone your doctor or Poisons Information Centre: 131126 (Australia) and 0800 764 766 (New Zealand) or go to the Accident and Emergency department at your nearest hospital if you think you or anyone else may have taken too many BIKTARVY tablets. Do this even if there are no signs of discomfort or poisoning. This may need urgent medical attention.
While you are taking BIKTARVY
Things you must not do
Do not breastfeed. See “Before you start to take it”
Avoid doing things that can spread HIV infection.
- Do not share needles or other injection equipment.
- Do not share personal items that can have blood or body fluids on them, like toothbrushes or razor blades.
Always practice safer sex by using a latex or polyurethane condom or other barrier to reduce the chance of sexual contact with semen, vaginal secretions, or blood.
Do not take BIKTARVY after the expiry or “use by” date (EXP) printed on the bottle. If you take it after the expiry date has passed, it may not work as well.
Do not take BIKTARVY if the packaging is torn or shows signs of tampering.
Things to be careful of
Be careful driving or operating machinery until you know how BIKTARVY affects you. If you are dizzy, have trouble concentrating, or are drowsy, avoid activities that may be dangerous, such as driving or operating machinery.
Side Effects
Like all medicines, BIKTARVY can have side effects, although not everybody gets them. Some may be serious and need medical attention.
Check with your doctor as soon as possible if you have any problems while taking BIKTARVY, even if you do not think the problems are connected with the medicine or are not listed in this leaflet.
Hepatic Flares
If you have both HIV infection and HBV infection you should not stop your BIKTARVY treatment without first discussing this with your doctor. Your HBV may get worse (flare-up) if you stop taking BIKTARVY. A “flare-up” or “hepatic flare” is when your HBV infection suddenly returns in a worse way than before. You may require medical exams and blood tests for several months after stopping treatment.
Signs and symptoms of inflammation
In some patients with advanced HIV infection (AIDS), signs and symptoms of inflammation from previous infections may occur soon after anti-HIV treatment is started. It is believed that these symptoms are due to an improvement in the body’s immune response, which lets the body fight infections that may have been present with no obvious symptoms. If you notice any symptoms of infection, please tell your doctor immediately.
Allergy
Some people are allergic to medicines. If you have any of the following symptoms soon after taking your medicine, DO NOT TAKE ANY MORE BIKTARVY and tell your doctor IMMEDIATELY or go to the accident and emergency department at your nearest hospital:
- skin troubles such as lumpy skin rash or “hives”
- swelling of the face, lips, mouth or throat which may cause difficulty in swallowing or breathing
- wheezing, chest pain or tightness
- fainting
These are very serious effects. If you have them, you may have a serious allergic reaction. You may need urgent medical attention or hospitalisation. Hypersensitivity reactions are very rare.
Common side effects
The most common side effects of BIKTARVY are diarrhoea and headache.
Other side effects include:
- nausea
- tiredness (fatigue)
- abdominal pain
- indigestion
- wind (flatulence)
- rash
- vomiting
- abnormal dreams
- suicidal behaviour
- anxiety
- sleep disorders
- depression
Talk to your doctor or pharmacist if you don’t understand anything in this list.
This is not a complete list of side effects possible with BIKTARVY.
Ask your doctor or pharmacist for a more complete list of side effects of BIKTARVY and all the medicines you will take.
After taking BIKTARVY
Storage
Keep BIKTARVY tablets where children cannot reach them. A locked cupboard at least one-and-a half metres above the ground is a good place to store them.
Keep BIKTARVY tablets in a cool, dry place where it stays below 30 °C.
Do not store BIKTARVY or any other medicine in a bathroom or near a sink.
Do not leave BIKTARVY in the car or on a window sill. Heat and dampness can destroy some medicines.
Keep your BIKTARVY tablets in the bottle with the cap tightly closed until you take them. If you take BIKTARVY tablets out of their pack they may not keep well.
Product Description
What the tablets look like
The 50/200/25 mg BIKTARVY tablets are capsule-shaped, purplish-brown in colour and film-coated.
Each tablet is debossed with “GSI” on one side and the number “9883” on the other side.
BIKTARVY tablets are supplied in bottles containing 30 tablets.
Ingredients
Each BIKTARVY tablet contains the active ingredients:
- bictegravir (as sodium)
- emtricitabine
- tenofovir alafenamide
Each BIKTARVY tablet also contains the following inactive ingredients:
- microcrystalline cellulose
- croscarmellose sodium
- magnesium stearate
Film-coating
- polyvinyl alcohol
- titanium dioxide
- macrogol 3350
- purified talc
- Opadry II Brown
Sponsor
BIKTARVY tablets are supplied in Australia by:
Gilead Sciences Pty Ltd
Level 6, 417 St Kilda Road
Melbourne, Victoria 3004
Date of preparation: 18 September 2020
BIKTARVY 50/200/25 mg tablets AUST R 291923
BIKTARVY, DESCOVY, GENVOYA, , ODEFSEY, TRUVADA, VEMLIDY and VIREAD are trademarks of Gilead Sciences, Inc., or its related companies. Other brands listed are trademarks of their respective owners and are not trademarks of Gilead Sciences, Inc.
Published by MIMS December 2022




 Additional adverse reactions occurring in less than 3% of subjects administered Biktarvy in Studies 1489 and 1490 included abnormal dreams, vomiting, flatulence, dyspepsia, abdominal pain, and rash. The majority of adverse reactions occurred at severity Grade 1. No additional adverse reactions were identified in the open-label extension phases of Studies 1489 and 1490.
Additional adverse reactions occurring in less than 3% of subjects administered Biktarvy in Studies 1489 and 1490 included abnormal dreams, vomiting, flatulence, dyspepsia, abdominal pain, and rash. The majority of adverse reactions occurred at severity Grade 1. No additional adverse reactions were identified in the open-label extension phases of Studies 1489 and 1490.
 Biktarvy was noninferior in achieving HIV-1 RNA < 50 copies/mL at both Week 48 and 144 when compared to ABC/DTG/3TC and to DTG+FTC/TAF, respectively. Treatment outcomes were similar among treatment groups across subgroups by age, sex, race, baseline viral load, and baseline CD4+ cell count.
Biktarvy was noninferior in achieving HIV-1 RNA < 50 copies/mL at both Week 48 and 144 when compared to ABC/DTG/3TC and to DTG+FTC/TAF, respectively. Treatment outcomes were similar among treatment groups across subgroups by age, sex, race, baseline viral load, and baseline CD4+ cell count. In Study 1844, at Week 48, switching to Biktarvy was noninferior to remaining on ABC/DTG/3TC. The percentages of patients with HIV-1 RNA ≥ 50 copies/mL and who maintained HIV-1 RNA < 50 copies/mL were similar between the Biktarvy and ABC/DTG/3TC groups. Treatment outcomes between treatment groups were similar across subgroups by age, sex and race. The mean change from baseline in CD4+ count at Week 48 was -31 cells/mm3 in patients who switched to Biktarvy and 4 cells/mm3 in patients who stayed on ABC/DTG/3TC.
In Study 1844, at Week 48, switching to Biktarvy was noninferior to remaining on ABC/DTG/3TC. The percentages of patients with HIV-1 RNA ≥ 50 copies/mL and who maintained HIV-1 RNA < 50 copies/mL were similar between the Biktarvy and ABC/DTG/3TC groups. Treatment outcomes between treatment groups were similar across subgroups by age, sex and race. The mean change from baseline in CD4+ count at Week 48 was -31 cells/mm3 in patients who switched to Biktarvy and 4 cells/mm3 in patients who stayed on ABC/DTG/3TC. The multiple dose pharmacokinetic parameters of the components of Biktarvy are provided in Table 10.
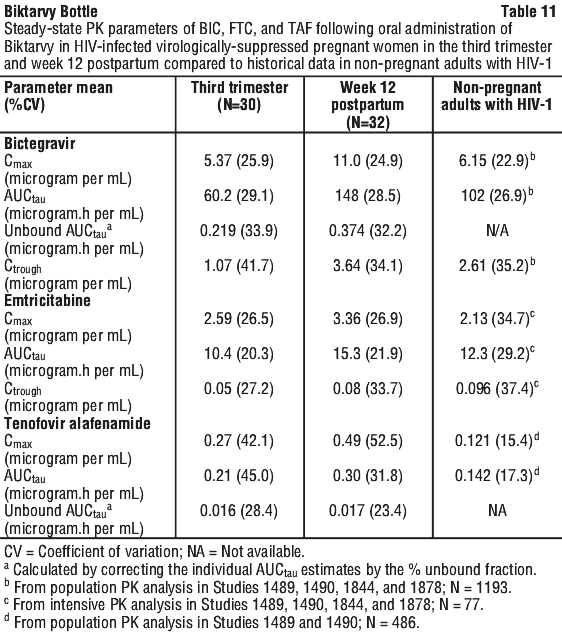
The multiple dose pharmacokinetic parameters of the components of Biktarvy are provided in Table 10.
 Bictegravir sodium is a white to off-white to yellow solid with a solubility of 0.1 mg/mL in water at 20°C.
Bictegravir sodium is a white to off-white to yellow solid with a solubility of 0.1 mg/mL in water at 20°C. FTC is a white to off-white crystalline powder with a solubility of approximately 112 mg/mL in water at 25°C. The partition coefficient (log p) for FTC is -0.43 and the pKa is 2.65.
FTC is a white to off-white crystalline powder with a solubility of approximately 112 mg/mL in water at 25°C. The partition coefficient (log p) for FTC is -0.43 and the pKa is 2.65. TAF is a white to off-white or tan powder with a solubility of 4.7 mg/mL in water at 20°C.
TAF is a white to off-white or tan powder with a solubility of 4.7 mg/mL in water at 20°C.