What is in this leaflet
This leaflet answers some common questions about bivalirudin. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you using this medicine against the benefits they expect it will have for you.
If you have any concerns about receiving this medicine, ask your doctor or pharmacist.
Keep this leaflet with the medicine. You may want to read it again.
What this medicine is used for
Bivalirudin helps prevent unwanted blood clotting during an angioplasty. Bivalirudin belongs to a group of medicines called anticoagulants.
How it works
An angioplasty is a medical procedure in which blocked blood vessels in the heart are unblocked. Angioplasty is also called percutaneous coronary intervention, or PCI. Angioplasty improves blood flow in the heart, helping heart problems such as angina.
Bivalirudin prevents blood clotting during an angioplasty by stopping thrombin from working. Thrombin is a protein that starts blood clotting.
Ask your doctor if you have any questions about why this medicine has been prescribed for you. Your doctor may have prescribed this medicine for another reason.
This medicine is not addictive.
This medicine is available only with a doctor's prescription.
There is not enough information to recommend the use of this medicine in children.
Before you are given this medicine
When you must not be given it
Do not receive this medicine if you have an allergy to:
- bivalirudin
- any of the ingredients listed at the end of this leaflet.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- tightness of the chest, wheezing or difficulty breathing
- swelling of the face, lips, tongue, throat or other parts of the body
- rash, itching or hives on the skin
Do not receive this medicine if you have or have had any of the following medical conditions:
- active bleeding or increased risk of bleeding because of haemostasis disorders or irreversible coagulation disorders
- severe uncontrolled hypertension
- subacute bacterial endocarditis
- severe kidney problems (such as kidney failure) or are on dialysis
Do not receive this medicine after the expiry date printed on the pack or if the packaging is torn or shows signs of tampering. If it has expired or is damaged, the pharmacist will arrange for its disposal.
If you are not sure whether you should receive this medicine, talk to your doctor.
Before you are given it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have or have had any of the following medical conditions:
- tend to bleed or bruise easily
- kidney or liver disease
- uncontrolled high blood pressure
- other heart conditions
Tell your doctor if you are already taking any medications such as blood-thinners/anticoagulants or medicines to prevent blood clots. Your risk of bleeding may increase. Your doctor will discuss with you the risks and benefits involved.
Tell your doctor if you are pregnant, plan to become pregnant or if you have recently given birth. Your risk of bleeding may increase. Your doctor will discuss with you the risks and benefits involved.
Tell your doctor if you are breastfeeding. It is not known whether this medicine passes into human breast milk. Your doctor will discuss with you the risks and benefits involved.
If you have not told your doctor about any of the above, tell them before you start receiving this medicine.
Taking other medicines
Tell your doctor and pharmacist if you are taking other medicines, including any that you get without a prescription from your pharmacy, health food shop or supermarket.
Some medicines may interact with bivalirudin. These include:
- other medicines use to stop blood clotting (e.g. warfarin, heparin)
- medicines which affect platelets (e.g. aspirin, ticlopidine, clopidogrel)
- St John's Wort, ginseng and ginkgo biloba
These medicines may be affected by bivalirudin or may affect how well it works. You may need different amounts of your medicines, or you may need to take different medicines.
Your doctor and pharmacist have more information on medicines to be careful with or avoid while taking this medicine.
How this medicine is given
Your doctor will give you bivalirudin into a vein as an injection or as a continuous infusion.
During angioplasty, bivalirudin should be given with aspirin.
How much is given
Your doctor will decide how much of this medicine you will receive.
If you have kidney disease, your doctor may change the usual dose.
If you receive too much (overdose)
As bivalirudin is given to you in a hospital under the supervision of your doctor or medical staff, it is very unlikely that you will receive an overdose.
You will be closely monitored while in the hospital so that any unwanted side effects can be treated, and you may need to stay overnight if you have certain medical conditions.
However, tell your doctor immediately if you experience side effects.
While you are using this medicine
Things you must do
If you are about to be started on any new medicine, remind your doctor and pharmacist that you are receiving this medicine.
Tell any other doctors, dentists, and pharmacists who treat you that you are receiving this medicine.
If you are going to have surgery, tell the surgeon or anaesthetist that you are receiving this medicine. Bivalirudin may affect other medicines used during surgery.
If you become pregnant or start to breastfeed while receiving this medicine, tell your doctor immediately.
If you are about to have any blood tests, tell your doctor that you are receiving this medicine. It may interfere with the results of some tests.
If you are taking warfarin, your doctor may do some blood clotting tests after your treatment with bivalirudin, as it may affect your INR levels.
Keep all your doctor's appointments so that your progress can be checked. Your doctor may do some tests from time to time to make sure the medicine is working and to prevent unwanted side effects.
Things you must not do
Do not give your medicine to anyone else, even if they have the same condition as you.
Side effects
Tell your doctor and nurse as soon as possible if you do not feel well after you have been given bivalirudin.
All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical attention if you get some of the side effects.
Do not be alarmed by the following lists of side effects. You may not experience any of them.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice any of the following:
- nausea or vomiting
- headache
- trouble falling asleep
- high or low blood pressure
- fever
- pain at injection site, chest or back
- slow or fast heart beat
- skin rash
- anaemia, decreased platelets or blood vessel disorders
If any of the following happen, tell your doctor and nurse immediately:
- bleeding from other parts of the body, such as at needle puncture wounds, in the urine, or bruising where the intravenous catheter was inserted
- shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue, throat or other parts of the body; tightness of the chest; rash, itching or hives on the skin (signs of an allergic reaction)
These may be serious side effects and you may need medical attention.
Tell your doctor or nurse if you notice anything that is making you feel unwell.
Other side effects not listed above may occur in some patients.
Storage and disposal
Storage
This medicine will be stored in the pharmacy or on the ward.
It is kept in a cool dry place where the temperature stays below 25°C. It should be stored in the original container.
After bivalirudin powder has been dissolved, use the solution as soon as possible.
Disposal
Bivalirudin is used for one dose in one patient only. Any remaining contents should be discarded.
Product description
What it looks like
White to off-white lyophilisate powder for reconstitution for IV injection. Supplied in a clear glass vial. AUST R 241714.
Ingredients
Each vial contains 250 mg of bivalirudin (as trifluoroacetate) as the active ingredient.
It also contains the following:
- mannitol
- sodium hydroxide
This medicine does not contain gluten, lactose, sucrose, tartrazine or any other azo dyes.
Sponsor
Arrotex Pharmaceuticals Pty Ltd
15 – 17 Chapel Street
Cremorne VIC 3121
This leaflet was prepared in July 2022.
Published by MIMS June 2023

 All parenteral drug products should be inspected visually for particulate matter and discolouration prior to administration. If particulate matter is observed in preparations of the parenteral drug products, these products should not be used with bivalirudin.
All parenteral drug products should be inspected visually for particulate matter and discolouration prior to administration. If particulate matter is observed in preparations of the parenteral drug products, these products should not be used with bivalirudin. Adverse drug events occurring in less than 1% of the ACUITY study population administered heparin plus GP IIb/IIIa inhibitor, bivalirudin plus GP IIb/IIIa inhibitor, or bivalirudin alone include bradycardia, haemorrhage, vascular pseudoaneurysm, rash, urticaria, and injection site reactions.
Adverse drug events occurring in less than 1% of the ACUITY study population administered heparin plus GP IIb/IIIa inhibitor, bivalirudin plus GP IIb/IIIa inhibitor, or bivalirudin alone include bradycardia, haemorrhage, vascular pseudoaneurysm, rash, urticaria, and injection site reactions. Both minor and major bleeds were significantly less frequent with bivalirudin than the heparin plus GP IIb/IIIa inhibitor comparator group. Major bleeding occurred most frequently at the sheath puncture site (see Table 4). Other less frequently observed bleeding sites with greater than 0.1% (uncommon) bleeding included "other" puncture site, retroperitoneal, gastrointestinal, ear, nose or throat.
Both minor and major bleeds were significantly less frequent with bivalirudin than the heparin plus GP IIb/IIIa inhibitor comparator group. Major bleeding occurred most frequently at the sheath puncture site (see Table 4). Other less frequently observed bleeding sites with greater than 0.1% (uncommon) bleeding included "other" puncture site, retroperitoneal, gastrointestinal, ear, nose or throat. Thrombocytopenia was reported in 10 bivalirudin-treated patients participating in the ACUITY study (0.1%). The majority of these patients received concomitant acetylsalicylic acid and clopidogrel, and 6 out of the 10 patients also received a GP IIb/IIIa inhibitor. Mortality among these patients was nil.
Thrombocytopenia was reported in 10 bivalirudin-treated patients participating in the ACUITY study (0.1%). The majority of these patients received concomitant acetylsalicylic acid and clopidogrel, and 6 out of the 10 patients also received a GP IIb/IIIa inhibitor. Mortality among these patients was nil. Adverse drug events occurring in less than 1% of the REPLACE-2 study population administered heparin plus GP IIb/IIIa inhibitor or bivalirudin plus provisional GP IIb/IIIa inhibitor include anaemia, allergic reaction, thrombosis, haemorrhage, vascular disorder, vascular anomaly, dyspnoea, rash, and injection site haemorrhage.
Adverse drug events occurring in less than 1% of the REPLACE-2 study population administered heparin plus GP IIb/IIIa inhibitor or bivalirudin plus provisional GP IIb/IIIa inhibitor include anaemia, allergic reaction, thrombosis, haemorrhage, vascular disorder, vascular anomaly, dyspnoea, rash, and injection site haemorrhage.

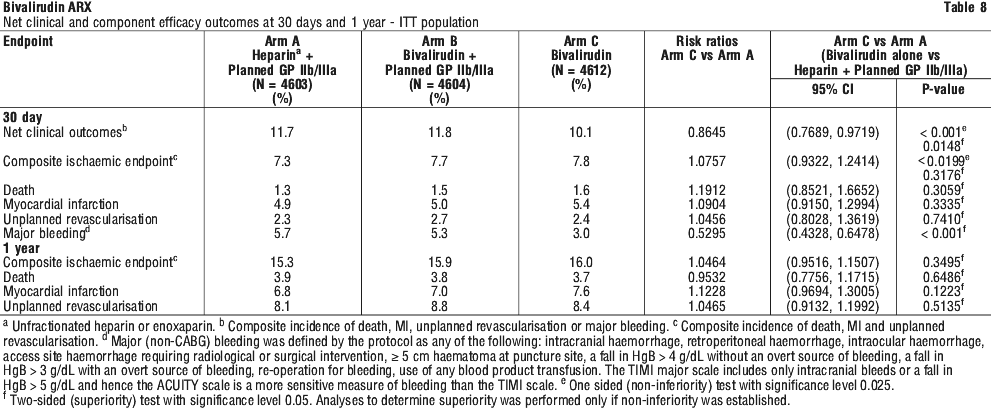 For patients that received aspirin and clopidogrel as per protocol (Table 9), at 30 days, use of bivalirudin alone was superior to heparins plus planned GP IIb/IIIa inhibitor with regards to major bleeding (ACUITY scale) (3.1% vs 5.9%, Psup < 0.0001) and for the net clinical endpoint (9.5% vs 11.8%, Psup = 0.0052).
For patients that received aspirin and clopidogrel as per protocol (Table 9), at 30 days, use of bivalirudin alone was superior to heparins plus planned GP IIb/IIIa inhibitor with regards to major bleeding (ACUITY scale) (3.1% vs 5.9%, Psup < 0.0001) and for the net clinical endpoint (9.5% vs 11.8%, Psup = 0.0052).
 At 12 months follow-up, mortality was 1.9% amongst patients randomised to bivalirudin plus provisional glycoprotein (GP) IIb/IIIa inhibitors and 2.5% amongst patients randomised to heparin plus planned GP IIb/IIIa inhibitors.
At 12 months follow-up, mortality was 1.9% amongst patients randomised to bivalirudin plus provisional glycoprotein (GP) IIb/IIIa inhibitors and 2.5% amongst patients randomised to heparin plus planned GP IIb/IIIa inhibitors.

 Glomerular filtration may contribute to clearance of intact bivalirudin peptide. Bivalirudin does not bind to plasma proteins (other than thrombin) or to red blood cells. Bivalirudin is haemodialysable.
Glomerular filtration may contribute to clearance of intact bivalirudin peptide. Bivalirudin does not bind to plasma proteins (other than thrombin) or to red blood cells. Bivalirudin is haemodialysable. All parenteral drug products should be inspected visually for particulate matter and discolouration prior to administration. If particulate matter is observed in preparations of the parenteral drug products, these products should not be used with bivalirudin.
All parenteral drug products should be inspected visually for particulate matter and discolouration prior to administration. If particulate matter is observed in preparations of the parenteral drug products, these products should not be used with bivalirudin.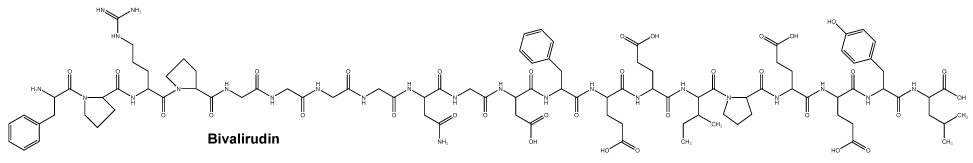 Chemical name: D-phenylalanyl-L-prolyl-L-arginyl-L-prolyl-glycyl-glycyl-glycyl-glycyl-L-asparagyl-glycyl-L-α-aspartyl-L-phenylalanyl-L-α-glutamyl-L-α-glutamyl-L-isoleucyl-L-prolyL-L-α-glutamyl-L-α-glutamyl-L-tyrosyl-L-leucine.
Chemical name: D-phenylalanyl-L-prolyl-L-arginyl-L-prolyl-glycyl-glycyl-glycyl-glycyl-L-asparagyl-glycyl-L-α-aspartyl-L-phenylalanyl-L-α-glutamyl-L-α-glutamyl-L-isoleucyl-L-prolyL-L-α-glutamyl-L-α-glutamyl-L-tyrosyl-L-leucine.