What is in this leaflet
This leaflet answers some common questions about Blincyto. It does not contain all the available information. It does not take the place of talking to your doctor, nurse or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you or your child taking Blincyto against the benefits they expect it will have for you.
If you have any concerns about taking this medicine, ask your doctor or pharmacist.
Keep this leaflet. You may need to read it again.
What Blincyto is used for
The active ingredient in Blincyto is blinatumomab. This belongs to a group of medicines called antineoplastic agents which target cancer cells.
Blincyto is used to treat adults, adolescents, and children with acute lymphoblastic leukaemia. Acute lymphoblastic leukaemia is a cancer of the blood in which a particular type of white blood cell is growing out of control. This medicine works by enabling your immune system to attack and destroy these abnormal white blood cancer cells.
Ask your doctor if you have any questions about why it has been prescribed for you.
This medicine is available only with a doctor's prescription.
Before you are given Blincyto
When you must not be given it
Do not receive Blincyto if you have an allergy to:
- any medicine containing blinatumomab.
- any of the ingredients listed at the end of this leaflet.
- any medicines that are produced using Chinese Hamster Ovary cells.
Some of the symptoms of an allergic reaction may include:
- shortness of breath
- wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
- rash, itching or hives on the skin.
Do not use it after the expiry date printed on the pack. If you use it after the expiry date has passed it may not work as well.
Do not use it if the packaging is torn or shows signs of tampering.
If you are not sure whether you should start taking this medicine, talk to your doctor, nurse or pharmacist.
Before you are given it
Tell your doctor if you have allergies to any other medicines, foods, preservatives or dyes.
Tell your doctor if you have any of the following medical conditions:
- if you have ever had neurological problems (such as seizures, memory loss, confusion, disorientation, loss of balance or difficulty in speaking)
- an active infection.
Tell your doctor if you have ever had an infusion reaction after previously using Blincyto.
Tell your doctor if you are pregnant, think you are pregnant, or plan to become pregnant. The effects of Blincyto in pregnant women are not known.
Women who are able to become pregnant should use contraception during treatment. You must also do this for 48 hours after your last treatment.
Talk to your doctor or nurse about suitable methods of contraception.
Tell your doctor if you are breastfeeding or planning to breast feed. It is not known whether Blincyto passes into breast milk.
You should not breast-feed during treatment with Blincyto and for at least 48 hours after your last treatment.
If you have not told your doctor about any of the above, tell them before you are given Blincyto.
Taking other medicines
Tell your doctor or nurse if you are taking any other medicines, including any that you get without a prescription from your pharmacy, supermarket or health food shop. These medicines may be affected by Blincyto or may affect how well it works. You may need to use different amounts of your medicines or take different medicines. Your doctor will advise you.
Tell your doctor or nurse if you think you may need any vaccinations in the near future, including those needed to travel to other countries. Some vaccines must not be given within two weeks before, at the same time as or in the months after you receive treatment with Blincyto.
Your doctor will check if you should have the vaccination.
How Blincyto is given
Blincyto is given to you through a vein (intravenous) continuously for 4 weeks by an infusion pump. This is followed by a 2-week break during which you will not be given the infusion. Each treatment cycle is 6 weeks. After this 2-week break, your doctor will determine if you will be given additional treatment cycles.
Your doctor or nurse will give you the infusion.
Detailed instructions for your doctor, nurse and pharmacist on how to prepare Blincyto are included in the Product Information. Your doctor will determine when your Blincyto infusion bag or cassette will be changed, which may range from every day to every 4 days. The infusion rate may be faster or slower depending on how often the bag or cassette is changed.
Medicines given before each treatment cycle
Before your treatment with Blincyto, you will be given other medicines (pre-medication) to help reduce reactions to the medication and a potentially life-threatening complication known as tumour lysis syndrome, which is caused by chemical disturbances in the blood due to the breakdown of dying cancer cells. These medicines may include corticosteroids.
Treatment of relapsed or refractory acute lymphoblastic leukaemia
It is recommended that the first 9 days of your first treatment cycle and the first 2 days of your second cycle with Blincyto are given to you in a hospital or clinic. This will allow you to be under the supervision of a doctor or nurse who can check you for side effects.
Patients greater than or equal to 45 kg body weight
- Your first cycle
For the first week of Blincyto treatment, your infusion pump will be set to deliver a dose of 9 micrograms per day. Your dose of Blincyto should be increased to 28 micrograms per day for weeks 2, 3 and 4 of your treatment depending on how you respond to treatment with Blincyto. - Your next cycles
If your doctor determines that more cycles of Blincyto should be given, your pump will be set to deliver a dose of 28 micrograms per day.
You may not be able to tell the difference between the 9 micrograms per day and 28 micrograms per day infusion bags.
Patients less than 45 kg body weight
The dose that your pump will be set to deliver is based on your weight.
- Your first cycle
After the first week of Blincyto treatment, the dose of Blincyto should be increased for weeks 2, 3, and 4 of your first cycle depending on how you respond to treatment with Blincyto. - Your next cycles
Your doctor will determine if more cycles of Blincyto should be given.
You may not be able to tell the difference between the dose delivered during the first week of your first cycle and the increased dose delivered for the remainder of the first cycle and for the subsequent cycles.
Treatment of Minimal Residual Disease positive ALL
It is recommended that the first 3 days of your first treatment cycle and the first two days of your second treatment cycle with Blincyto be given to you in a hospital or clinic. This will allow you to be under the supervision of a doctor or nurse who can check you for side effects.
Patients greater than or equal to 45 kg body weight
- All treatment cycles
Your doctor will determine the number of cycles of Blincyto that should be given. Your pump will be set to deliver a dose of 28 micrograms per day.
Infusion pump and intravenous tubing
Your doctor or nurse will advise you on how to manage your daily activities around your infusion pump.
How long to take it
The number of treatment cycles and the dose that you will be given will depend on how well you tolerate and respond to Blincyto. Your doctor will discuss with you how long your treatment will last. Your treatment may be interrupted depending on how well you tolerate Blincyto.
While you are using Blincyto
Things you must do
Keep the area around the catheter clean if you have a catheter for infusion. This is very important, otherwise you could get an infection.
Your doctor or nurse will show you how to care for your catheter site.
If there is a problem with the infusion pump or the pump alarm sounds, do not adjust the settings on the pump. Any changes to the pump settings may result in a dose that is too high or too low.
Tell your doctor or nurse immediately if:
- there is a problem with the pump or if the pump alarm sounds
- the infusion bag empties before the scheduled bag change
- the infusion pump stops unexpectedly.
Do not try to restart the pump.
Tell your doctor, nurse or pharmacist that you are taking Blincyto before you start any new medicine.
Tell any other doctors who treat you that you are taking this medicine.
Tell your doctor, nurse or pharmacist immediately if you experience any of the following while being treated with Blincyto:
- seizures, difficulty in speaking or slurred speech, confusion and disorientation, or loss of balance
- chills or shivering, or feel warm
Take your temperature as you may have a fever - these may be symptoms of an infection. - a reaction at any time during your infusion. Symptoms may include dizziness, face swelling, difficulty breathing, wheezing, or rash.
- severe and persistent stomach pain, with or without nausea and vomiting. These may be symptoms of a serious and potentially fatal condition known as pancreatitis (inflammation of the pancreas).
The reaction may have to be treated and your dose of Blincyto may need to be adjusted.
Your doctor or nurse will monitor you for signs and symptoms of these reactions.
You may experience a severe low white blood cell count or severe low white blood count with fever (neutropenia or febrile neutropenia) or elevated liver enzymes.
Your doctor or nurse will take regular blood tests to monitor your blood counts during treatment with Blincyto.
If you become pregnant while taking this medicine, tell your doctor immediately. Your doctor may need to talk to you about precautions in using vaccinations for your baby.
Some side effects more frequently seen in adolescents and children include:
- runny nose (rhinitis)
- nose bleeds.
Tell your doctor or nurse if you think you may need any vaccinations in the near future, including those needed to travel to other countries. Some vaccines must not be given within two weeks before, at the same time as or in the months after you receive treatment with Blincyto.
Your doctor will check if you should have the vaccination.
Things you must not do
Do not drive, use heavy machines, or engage in hazardous activities while you are being given Blincyto. Blincyto can cause problems such as dizziness, seizures, and confusion.
If you take too much (overdose)
Immediately telephone your doctor for advice or go to Accident and Emergency at the nearest hospital, if you think that you may have taken too much Blincyto.
Do this even if there are no signs of discomfort or poisoning. You may need urgent medical attention.
Side effects
Tell your doctor, nurse or pharmacist as soon as possible if you do not feel well while you are being given Blincyto.
All medicines can have side effects. You may need medical attention if you get some of the side effects.
Ask your doctor, nurse or pharmacist to answer any questions you may have.
Tell your doctor, nurse or pharmacist if you notice any of the following:
- tingling skin
- rapid heart rate
- cough
- nausea, constipation, diarrhoea, stomach pain, vomiting
- rash
- back pain, pain in hands or feet, painful/swollen joints, bone pain
- swollen hands, ankles or feet
- flushing
- tiredness
- weight gain.
The above list includes the more common side effects of your medicine.
Tell your doctor immediately if any of the following happen:
- fever, swelling or chills, shortness of breath, headache and dizziness any of which may become severe
- difficulty sleeping (insomnia)
- confusion, disorientation
- headache, shaking (tremor), dizziness
- difficulty in speaking, communicating, thinking or processing thoughts, or remembering things
- seizures (convulsions)
- chest pain or other pain
- swelling of the face, lips, mouth, tongue or throat which may cause difficulty in swallowing or breathing.
The above list includes very serious side effects. You may need urgent medical attention or hospitalisation.
Tell your doctor or nurse if you notice anything that is making you feel unwell.
Other side effects not listed above may also occur in some people.
Do not be alarmed by this list of side effects. You may not experience any of them.
After preparing it
Storage
Blincyto solution for infusion will be prepared by your pharmacist and stored in a refrigerator at 2°C to 8°C until infusion.
Once at room temperature (up to 25°C) the solution will be infused within 96 hours.
Disposal
Your doctor, nurse or pharmacist will dispose of this medicine.
Product description
What it looks like
Each pack of Blincyto contains:
- 1 single-use Blincyto vial containing a sterile, preservative free, white to off-white lyophilised powder
- 1 single-use IV Solution Stabiliser vial containing sterile, preservative-free, colourless-to-slightly yellow, clear solution.
Ingredients
Active ingredient: blinatumomab.
The other ingredients in the Blincyto vial are:
- citric acid monohydrate
- trehalose dihydrate
- lysine hydrochloride
- polysorbate 80
- sodium hydroxide
Ingredients in the IV Solution Stabiliser vial are:
- citric acid monohydrate
- lysine hydrochloride
- polysorbate 80
- sodium hydroxide
- water for injections
This medicine does not contain lactose, sucrose, gluten, tartrazine or any other azo dyes.
Sponsor
Blincyto is supplied in Australia by:
Amgen Australian Pty Ltd
ABN 31 051 057 428
Level 7, 123 Epping Rd
North Ryde NSW 2113
Medical Information: 1800 803 638
Email: [email protected]
® = Registered Trademark
AUST R 232805 Blincyto 38.5 microgram powder for injection vial.
This leaflet was prepared in July 2020.
Published by MIMS September 2020

 Intrathecal chemotherapy prophylaxis is recommended before and during Blincyto therapy to prevent central nervous system ALL relapse.
Intrathecal chemotherapy prophylaxis is recommended before and during Blincyto therapy to prevent central nervous system ALL relapse.







 The adverse reaction profile in Blincyto-treated patients in this study was similar in type to those seen in the phase 1/2 single-arm studies; capillary leak syndrome was observed in one patient in the phase 2 single-arm study (Study 2, MT103-211).
The adverse reaction profile in Blincyto-treated patients in this study was similar in type to those seen in the phase 1/2 single-arm studies; capillary leak syndrome was observed in one patient in the phase 2 single-arm study (Study 2, MT103-211).

 Blincyto was administered as a continuous intravenous infusion. In the first cycle, the initial dose was 9 micrograms/day for week 1, then 28 micrograms/day for the remaining 3 weeks. The target dose of 28 micrograms/day was administered in cycle 2 and subsequent cycles starting on day 1 of each cycle. Dose adjustment was possible in case of adverse events. Of the 267 patients who received Blincyto, the median number of treatment cycles was two (range: 0 to 9 cycles); of the 109 patients who received SOC chemotherapy, the median number of treatment cycles was one (range: 1 to 4 cycles).
Blincyto was administered as a continuous intravenous infusion. In the first cycle, the initial dose was 9 micrograms/day for week 1, then 28 micrograms/day for the remaining 3 weeks. The target dose of 28 micrograms/day was administered in cycle 2 and subsequent cycles starting on day 1 of each cycle. Dose adjustment was possible in case of adverse events. Of the 267 patients who received Blincyto, the median number of treatment cycles was two (range: 0 to 9 cycles); of the 109 patients who received SOC chemotherapy, the median number of treatment cycles was one (range: 1 to 4 cycles).

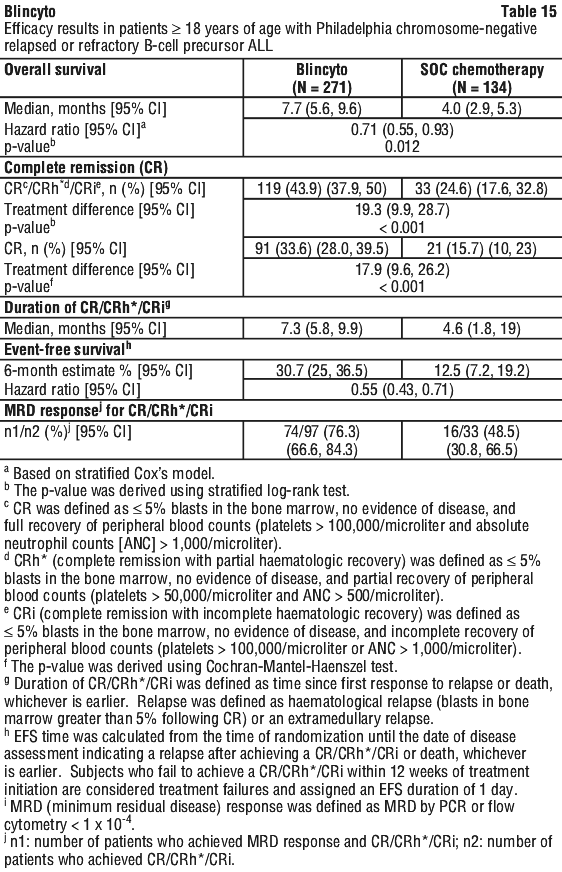 In Study 2 (MT103-211), the safety and efficacy of Blincyto were evaluated in an open-label, multicentre, single-arm study. Eligible patients were ≥ 18 years of age with Philadelphia chromosome-negative relapsed or refractory B-precursor ALL (relapsed with first remission duration of ≤ 12 months in first salvage or relapsed or refractory after first salvage therapy or relapsed within 12 months of allogeneic HSCT, and had ≥ 10% blasts in bone marrow).
In Study 2 (MT103-211), the safety and efficacy of Blincyto were evaluated in an open-label, multicentre, single-arm study. Eligible patients were ≥ 18 years of age with Philadelphia chromosome-negative relapsed or refractory B-precursor ALL (relapsed with first remission duration of ≤ 12 months in first salvage or relapsed or refractory after first salvage therapy or relapsed within 12 months of allogeneic HSCT, and had ≥ 10% blasts in bone marrow). Patients with prior allogeneic HSCT had similar response rates to those without prior HSCT, older patients had similar response rates to younger patients, and no substantial difference was observed in remission rates based on the number of lines of prior salvage treatment (see Figure 3).
Patients with prior allogeneic HSCT had similar response rates to those without prior HSCT, older patients had similar response rates to younger patients, and no substantial difference was observed in remission rates based on the number of lines of prior salvage treatment (see Figure 3). In Study 3 (MT103-206), the safety and efficacy of Blincyto were evaluated in an open-label, multicentre, dose escalation study in 36 patients (including 23 patients treated at a dose equivalent to the registrational dose) ≥ 18 years of age with relapsed and/or refractory B precursor ALL (first or greater relapse, refractory, or relapse after haematopoietic stem cell transplantation [HSCT]). Fifteen out of 36 (41.7%) patients had undergone allogeneic haematopoietic stem cell transplantation (HSCT) prior to receiving Blincyto. The complete remission/complete remission with partial haematological recovery (CR/CRh*) rate was 69.4% [25 out of 36 patients (95% CI: 51.9% - 83.7%): 15 (41.7%; 95% CI: 25.5% - 59.2%) CR; 10 (27.8%; 95% CI: 14.2% - 45.2%) CRh*]. Twenty two out of 25 (88%) patients with haematologic CR also had MRD responses (defined as MRD by PCR < 1 x 10-4). The median duration of remission was 8.9 months, and the median relapse-free survival (RFS) was 7.6 months. The median OS was 9.8 months.
In Study 3 (MT103-206), the safety and efficacy of Blincyto were evaluated in an open-label, multicentre, dose escalation study in 36 patients (including 23 patients treated at a dose equivalent to the registrational dose) ≥ 18 years of age with relapsed and/or refractory B precursor ALL (first or greater relapse, refractory, or relapse after haematopoietic stem cell transplantation [HSCT]). Fifteen out of 36 (41.7%) patients had undergone allogeneic haematopoietic stem cell transplantation (HSCT) prior to receiving Blincyto. The complete remission/complete remission with partial haematological recovery (CR/CRh*) rate was 69.4% [25 out of 36 patients (95% CI: 51.9% - 83.7%): 15 (41.7%; 95% CI: 25.5% - 59.2%) CR; 10 (27.8%; 95% CI: 14.2% - 45.2%) CRh*]. Twenty two out of 25 (88%) patients with haematologic CR also had MRD responses (defined as MRD by PCR < 1 x 10-4). The median duration of remission was 8.9 months, and the median relapse-free survival (RFS) was 7.6 months. The median OS was 9.8 months. The primary endpoint was the CR/CRh* rate within two cycles of treatment with Blincyto. Sixteen out of 45 (35.6%) patients achieved CR/CRh* within the first two treatment cycles. Of the 16 patients with CR/CRh* in the first 2 cycles, 12 of 14 (85.7%) patients with a CR and 2 of 2 (100%) patients with a CRh* also achieved an MRD complete response. See Table 18 for efficacy results from Study 4.
The primary endpoint was the CR/CRh* rate within two cycles of treatment with Blincyto. Sixteen out of 45 (35.6%) patients achieved CR/CRh* within the first two treatment cycles. Of the 16 patients with CR/CRh* in the first 2 cycles, 12 of 14 (85.7%) patients with a CR and 2 of 2 (100%) patients with a CRh* also achieved an MRD complete response. See Table 18 for efficacy results from Study 4. Treatment effects in evaluable subgroups (e.g. mutation status, number of prior TKIs, prior HSCT status, and relapse without prior HSCT) were in general consistent with the results in the overall population. Patients with T315I mutation, other mutations, or additional cytogenetic abnormalities responded with a similar rate as compared to those that did not have these mutations or abnormalities.
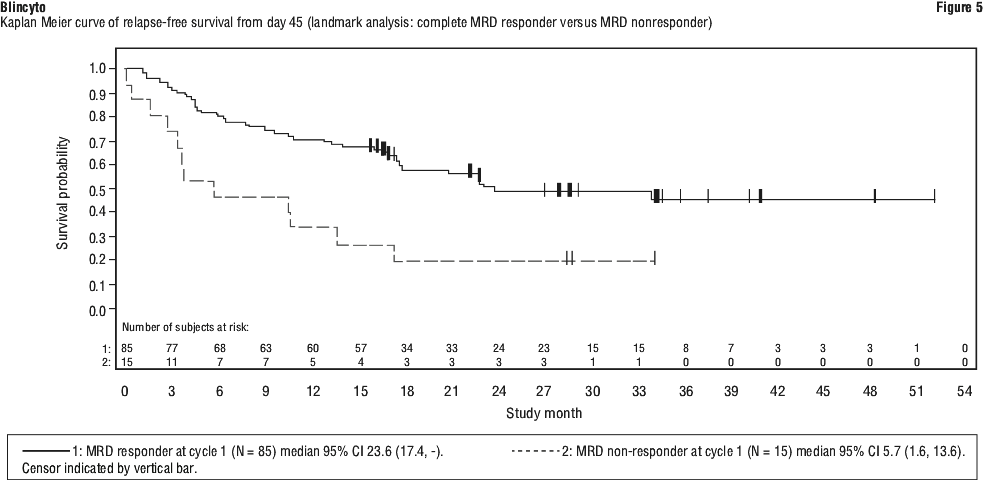
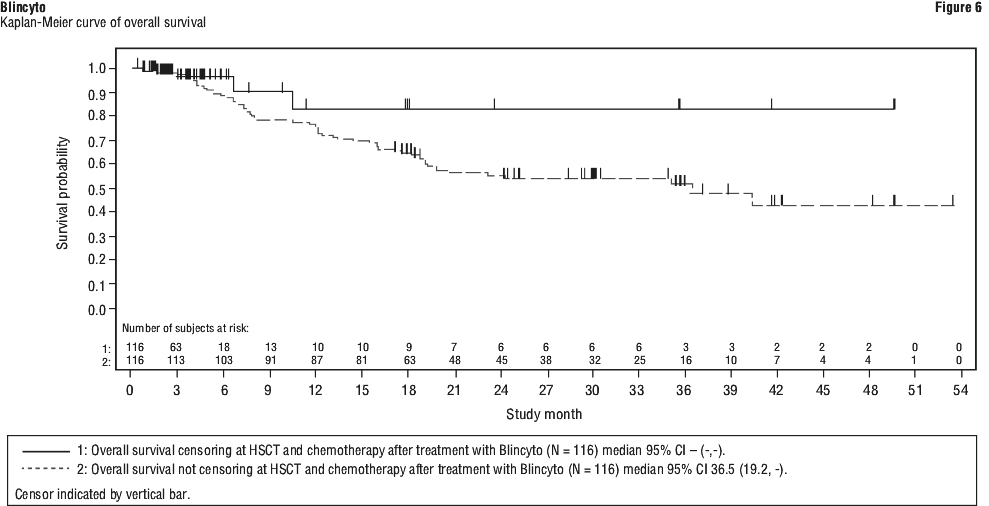
Treatment effects in evaluable subgroups (e.g. mutation status, number of prior TKIs, prior HSCT status, and relapse without prior HSCT) were in general consistent with the results in the overall population. Patients with T315I mutation, other mutations, or additional cytogenetic abnormalities responded with a similar rate as compared to those that did not have these mutations or abnormalities. The primary endpoint was the proportion of patients who achieved a complete MRD response within one cycle of Blincyto treatment. Eighty-eight out of 113 (77.9%) patients achieved a complete MRD response after one cycle of treatment. MRD response rates by age and MRD level at baseline subgroups were consistent with the results in the overall population. See Table 20 and Figures 4 to 6 for efficacy results from Study 5.
The primary endpoint was the proportion of patients who achieved a complete MRD response within one cycle of Blincyto treatment. Eighty-eight out of 113 (77.9%) patients achieved a complete MRD response after one cycle of treatment. MRD response rates by age and MRD level at baseline subgroups were consistent with the results in the overall population. See Table 20 and Figures 4 to 6 for efficacy results from Study 5.

 The primary endpoint was event-free survival (EFS). The study demonstrated statistically significant improvement in EFS for patients treated with Blincyto as compared to SOC consolidation chemotherapy. The overall median follow-up time for EFS was 51.9 months and the overall median follow-up time for the key secondary endpoint OS was 55.2 months. See Figure 7, Table 22, and Figure 8 for efficacy results from Study 6.
The primary endpoint was event-free survival (EFS). The study demonstrated statistically significant improvement in EFS for patients treated with Blincyto as compared to SOC consolidation chemotherapy. The overall median follow-up time for EFS was 51.9 months and the overall median follow-up time for the key secondary endpoint OS was 55.2 months. See Figure 7, Table 22, and Figure 8 for efficacy results from Study 6.

 In Study 7 (MT103-205) the safety and efficacy of Blincyto were evaluated in an open-label, multicentre, single-arm, phase 1/2 study in 93 paediatric patients with relapsed or refractory B cell precursor ALL (second or later bone marrow relapse, in any marrow relapse after allogeneic HSCT, or refractory to other treatments, and have > 25% blasts in bone marrow).
In Study 7 (MT103-205) the safety and efficacy of Blincyto were evaluated in an open-label, multicentre, single-arm, phase 1/2 study in 93 paediatric patients with relapsed or refractory B cell precursor ALL (second or later bone marrow relapse, in any marrow relapse after allogeneic HSCT, or refractory to other treatments, and have > 25% blasts in bone marrow). In Study 8 (AALL1331), the safety and efficacy of Blincyto were evaluated in a risk-stratified, randomised, Phase 3, open label study in paediatric and young adult patients (≥ 1 to < 31 years of age) at first relapse with childhood B-cell ALL. Enrolled patients received reinduction chemotherapy, and upon completion, were risk-assessed as either high risk (HR), intermediate risk (IR), low risk (LR) relapse, or treatment-failure. Risk stratification was based on site of relapse, time to relapse, and end-of-reinduction bone marrow morphology and MRD levels. The primary endpoint was disease-free survival (DFS). Blincyto was administered by continuous IV infusion at a dose of 15 microgram/m2/day. Each Blincyto cycle lasted 5 weeks (28-day continuous IV infusion, followed by a 7-day treatment-free interval). Thirty to sixty minutes before the start of therapy with Blincyto on Day 1 of cycle 1, patients were premedicated with 5 mg/m2 dexamethasone either orally or intravenously. Baseline demographics information is presented in Table 24.
In Study 8 (AALL1331), the safety and efficacy of Blincyto were evaluated in a risk-stratified, randomised, Phase 3, open label study in paediatric and young adult patients (≥ 1 to < 31 years of age) at first relapse with childhood B-cell ALL. Enrolled patients received reinduction chemotherapy, and upon completion, were risk-assessed as either high risk (HR), intermediate risk (IR), low risk (LR) relapse, or treatment-failure. Risk stratification was based on site of relapse, time to relapse, and end-of-reinduction bone marrow morphology and MRD levels. The primary endpoint was disease-free survival (DFS). Blincyto was administered by continuous IV infusion at a dose of 15 microgram/m2/day. Each Blincyto cycle lasted 5 weeks (28-day continuous IV infusion, followed by a 7-day treatment-free interval). Thirty to sixty minutes before the start of therapy with Blincyto on Day 1 of cycle 1, patients were premedicated with 5 mg/m2 dexamethasone either orally or intravenously. Baseline demographics information is presented in Table 24. In the HR/IR group, 103 eligible patients were randomised to receive chemotherapy and 105 eligible patients were randomised to receive 2 cycles of Blincyto. At the time of primary analysis, the median follow-up time for DFS in the HR/IR group was 3.4 years for the Blincyto arm. The 2-year DFS rate was 53.1% (95% CI: 43.1% to 62.1%) in the blinatumomab arm and 37.4% (95% CI: 27.7%, 47.0%) in the chemotherapy arm. The difference was not statistically significant (1-sided p = 0.027). The DFS hazard ratio from a stratified Cox proportional hazard model was 0.69 (95% CI: 0.47, 1.01). Randomisation of the HR/IR group of subjects was terminated early by the independent Data Safety Monitoring Committee (DSMC), based on a combined assessment of improved efficacy and safety in the Blincyto arm.
In the HR/IR group, 103 eligible patients were randomised to receive chemotherapy and 105 eligible patients were randomised to receive 2 cycles of Blincyto. At the time of primary analysis, the median follow-up time for DFS in the HR/IR group was 3.4 years for the Blincyto arm. The 2-year DFS rate was 53.1% (95% CI: 43.1% to 62.1%) in the blinatumomab arm and 37.4% (95% CI: 27.7%, 47.0%) in the chemotherapy arm. The difference was not statistically significant (1-sided p = 0.027). The DFS hazard ratio from a stratified Cox proportional hazard model was 0.69 (95% CI: 0.47, 1.01). Randomisation of the HR/IR group of subjects was terminated early by the independent Data Safety Monitoring Committee (DSMC), based on a combined assessment of improved efficacy and safety in the Blincyto arm. The primary endpoint was OS in patients who were MRD-negative. Secondary endpoints included RFS in patients who were MRD-negative, OS and RFS in patients who were MRD-positive.
The primary endpoint was OS in patients who were MRD-negative. Secondary endpoints included RFS in patients who were MRD-negative, OS and RFS in patients who were MRD-positive.

 The maximum storage time of the prepared IV bag at room temperature should not be longer than 6 hours prior to the start of infusion.
The maximum storage time of the prepared IV bag at room temperature should not be longer than 6 hours prior to the start of infusion. Using recombinant DNA technology, Blincyto is produced in a well-characterised mammalian cell (Chinese hamster ovary) culture and is purified by a series of steps that include measures to inactivate and remove viruses.
Using recombinant DNA technology, Blincyto is produced in a well-characterised mammalian cell (Chinese hamster ovary) culture and is purified by a series of steps that include measures to inactivate and remove viruses.