What is in this leaflet
This leaflet answers some common questions about Bortezomib Juno Powder for Injection. It does not contain all the available information. It does not take the place of talking to your doctor or pharmacist.
All medicines have risks and benefits. Your doctor has weighed the risks of you being given Bortezomib Juno against the benefits this medicine is expected to have for you.
If you have any concerns about being given Bortezomib Juno ask your doctor.
Keep this leaflet while being treated. You may need to read it again.
What Bortezomib Juno is used for
Bortezomib Juno belongs to a group of medicines called antineoplastic or cytotoxic medicines. You may also hear of these being called chemotherapy medicines. These medicines are used to kill cancer cells.
Bortezomib Juno is used to treat adults with multiple myeloma (cancer of the bone marrow). It is prescribed for patients who have not been previously treated for multiple myeloma. It is also prescribed for patients who have received one or more prior treatments and whose cancer is still progressing.
Bortezomib Juno is also used for the treatment of mantle cell lymphoma (a type of cancer affecting the lymph nodes) in adults in combination with the medicines rituximab, cyclophosphamide, doxorubicin and prednisone, for patients whose disease has not been previously treated.
Your doctor may have prescribed Bortezomib Juno for another reason.
Ask your doctor if you have any questions about why Bortezomib Juno has been prescribed for you.
This medicine is available only with a doctor's prescription.
Before you are given Bortezomib Juno
When you must use it
Do not use Bortezomib Juno if:
- you know you are allergic (hypersensitive) to bortezomib or boron or mannitol.
Symptoms of an allergic reaction may include rash, itching or hives on the skin, shortness of breath, wheezing or difficulty breathing, swelling of the face, lips, tongue or other parts of the body.
Before you start to use it:
Tell your doctor if you have or have had any medical conditions, especially the following:
- blood disorder with a low level of red or white blood cells or platelets. This disorder may become worse during treatment with Bortezomib Juno.
- if you are suffering from diarrhoea or vomiting as this may become worse during treatment with Bortezomib Juno.
- a history of fainting, dizziness or light-headedness.
- kidney problems
- liver problems, including hepatitis infection
- problems with numbness, tingling or pain in the hands or feet (neuropathy). This effect may be worsened by treatment with Bortezomib Juno.
- seizures
- any bleeding problems
- problems with your heart
- lung or breathing problems
Tell your doctor if you are pregnant or intend to become pregnant. Like most medicines used to treat cancer, Bortezomib Juno is not recommended for use during pregnancy.
Tell your doctor if you are breastfeeding or intend to breastfeed. It is not known whether Bortezomib Juno passes into breast milk. Therefore there is a possibility that the breastfed baby may be affected.
If you wish to restart breast-feeding after your Bortezomib Juno treatment, you must discuss this with your doctor or nurse, who will tell you when it is safe to do so.
Tell your doctor if you are trying to make your partner pregnant.
Both men and women receiving Bortezomib Juno and their partners must use a reliable method of contraception during and for 3 months after receiving Bortezomib Juno.
If you have not told your doctor about any of the above, tell them before you start treatment with Bortezomib Juno.
Taking other medicines:
Tell your doctor if you are taking any other medicines, including medicines you can buy without a prescription from a pharmacy, supermarket or health food shop.
In particular, tell your doctor if you are taking any of the following:
- amiodarone, a medicine used to treat irregular heart beat
- medicines used to treat viral infections such as flu, herpes and HIV
- isoniazid, a medicine used to treat tuberculosis
- nitrofurantoin, a medicine used to treat urinary tract infections
- ketoconazole, a medicine used to treat fungal infections
- ritonavir, a medicine used to treat HIV infection
- rifampicin, a medicine used to treat infections such as tuberculosis
- medicines used to treat high cholesterol levels in the blood
- medicines used to treat diabetes
- medicines that may lower blood pressure
- medicine used to treat epilepsy such as carbamazepine and phenobarbital
- phenytoin, a medicine used in preventing seizures
- St John's Wort (Hypericum perforatum).
These medicines may be affected by Bortezomib Juno or may affect how well Bortezomib Juno works. Your doctor or pharmacist can tell you what to do if you are using any of these medicines.
How Bortezomib Juno is given
Overall treatment with Bortezomib Juno must be done under the supervision of a doctor. Your treatment with Bortezomib Juno may be given by a healthcare professional (eg doctor or nurse) experienced in the administration of oncology medicines (see "How it is given").
How much is given
Your doctor will decide what dose you will receive. The dose will be calculated from your height and weight. It will also depend on factors such as kidney function, liver function and other medicines you are being given.
The safety of treatment with Bortezomib Juno in people with severe kidney function problems had not been well-studied.
The usual starting dose is 1.3 milligrams per square meter body surface area. Your doctor may change the dose during treatment depending on your response.
Ask your doctor if you want to know more about the dose of Bortezomib Juno you receive.
How it is given
Bortezomib Juno will be dissolved in sterile normal sodium chloride (salt) solution for injection. The solution is given as an injection into your vein (intravenously) over 3 to 5 seconds. The injection tube will be rinsed with a small quantity of sterile normal sodium chloride (salt) solution.
The solution can also be given subcutaneously as an injection into your thighs (right or left), or abdomen (right or left). Bortezomib Juno must be given intravenously or subcutaneously only. Bortezomib Juno must not be given into the space around the spinal cord (intrathecally).
When it is given
Multiple Myeloma
One cycle of treatment with Bortezomib Juno may consist of a total of 4 doses given over 3 weeks. Doses are given on days 1, 4, 8 and 11 followed by a ten day break from the treatment.
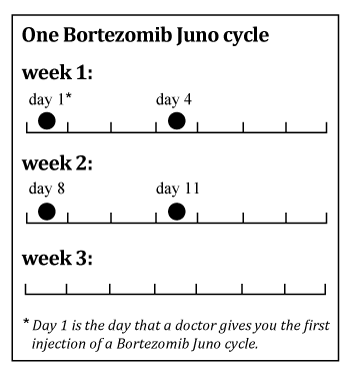
When Bortezomib Juno is given with thalidomide and dexamethasone, the treatment consists of a total of 3 cycles (9 weeks) for the induction stage. During the induction stage, Bortezomib Juno is administered twice weekly (days 1, 4, 8 and 11).
When Bortezomib Juno is given with dexamethasone, the treatment consists of a total of 4 cycles (12 weeks). Bortezomib Juno will be administered twice weekly (days 1, 4, 8 and 11).
When Bortezomib Juno is given with melphalan and prednisone, one cycle of treatment is 6 weeks and the treatment consists of a total of 9 cycles (54 weeks). In Cycles 1-4, Bortezomib Juno is administered twice weekly (days 1, 4, 8, 11, 22, 25, 29 and 32). In Cycles 5-9, Bortezomib Juno is administered once weekly (days (1, 8, 22 and 29).
Mantle Cell Lymphoma
When Bortezomib Juno is given with rituximab, cyclophosphamide, doxorubicin and prednisone, one cycle is 3 weeks and the treatment consists of a total of up to 8 cycles (24 weeks). For each cycle, Bortezomib Juno is given on days 1, 4, 8 and 11, followed by a ten day break from the treatment. Your doctor will decide on the number of cycles of Bortezomib Juno needed. This will depend on how you respond to treatment
If you take too much (overdose)
As Bortezomib Juno is given to you under the supervision of your doctor, it is very unlikely that you will receive too much. However, if you experience any side effects after being given Bortezomib Juno, tell you doctor or nurse immediately or go to Accident and Emergency at your nearest hospital. You may need urgent medical attention.
While you are being using Bortezomib Juno
Things you must do
Be sure to keep all your doctor's appointments so your progress can be checked. Your doctor will want to do some blood, urine and other tests from time to time to check on your progress and detect any unwanted side effects.
Keep follow up appointments with your doctor. It is important to have your follow-up doses of Bortezomib Juno at the appropriate times to get the best effects from your treatment.
Be sure to follow up your doctor's instructions about other medicines you should take, and other things you should do.
You may need to take other medicines to help prevent unwanted effects of Bortezomib Juno. You may also need to drink extra fluids if you experience vomiting and/or diarrhoea. Ask your doctor or pharmacist if you have any questions.
Tell any other doctors, dentists and pharmacists who are treating you that you are having Bortezomib Juno.
If you are about to be started on any new medicines, tell your doctor, dentist or pharmacist that you are having Bortezomib Juno.
If you plan to have surgery, tell your doctor or dentist that you are having Bortezomib Juno. If you become pregnant or your partner becomes pregnant while being given Bortezomib Juno, tell your doctor immediately.
Bortezomib Juno can lower the number of white blood cells and platelets in your blood. This means that you have an increased chance of getting an infection or bleeding. The following precautions should be taken to reduce your risk of infection or bleeding:
- Avoid people who have infections. Check with your doctor immediately if you think you may be getting an infection, or if you get a fever, chills, cough, hoarse throat, lower back or side pain or find it’s painful or difficult to urinate.
- Be careful when using a toothbrush, toothpick or dental floss. Your doctor, dentist, nurse or pharmacist may recommend other ways to clean your teeth and gums. Check with your doctor before having any dental work.
- Be careful not to cut yourself when you are using sharp objects such as a razor or nail cutters.
Things to be careful of
Be careful driving or operating machinery until you know how Bortezomib Juno affects you.
Bortezomib Juno may cause tiredness, light-headedness, dizziness, fainting, double or blurred vision in some people. Make sure you know how you react to Bortezomib Juno before you drive a car, operate machinery, or do anything else that could be dangerous if you are dizzy, light headed or have double or blurred vision. If you drink alcohol, dizziness or lightheadedness may be worse.
You may feel dizzy or faint when you get up quickly after sitting or lying down. Getting up slowly may help.
Side effects
Like all medicines, Bortezomib Juno can have side effects. Some of these effects may be serious. However there may be ways to reduce the discomfort of these effects. You may need medical treatment if you get some of the side effects.
Tell your doctor, nurse or pharmacist as soon as possible if you do not feel well while you are being treated with Bortezomib Juno.
Below is a list of the more common side effects you could get while being treated with Bortezomib Juno:
- tiredness, generally feeling unwell, weakness
- feeling sick (nausea) or vomiting
- diarrhoea
- constipation
- loss of appetite, and/or weight, fear of gaining weight
- bleeding or bruising more easily than normal
- sensitivity, numbness, tingling or burning sensation of the skin, or pain in the hands or feet
- fever, chills
- anaemia (a condition in which there is a decreased number of red blood cells)
- frequent infections such as fever, severe chills, sore throat or mouth ulcers
- herpes virus or hepatitis
- infections
- headache
- trouble sleeping, sweating,
- anxiety, mood swings, confusion or depression
- painful, swollen joints
- pain in your limbs, back pain, bone pain, muscle cramps
- swelling (around the eyes or in the ankles, wrists, arms, legs or face)
- pins and needles and unpleasant sensations
- difficulty in breathing
- dizziness
- dehydration
- cough
- aching muscles, muscle tenderness or weakness not caused by exercise
- uncomfortable feeling in the stomach or belching after eating
- stomach pain
- blockage in the intestine
- bad taste in the mouth
- low blood pressure (dizziness, light headedness or fainting)
- high blood pressure
- chest pain
- small blisters in clusters on the skin (herpes)
- rash, itching
- redness of the skin or redness and pain at injection site
- hair loss
- blurred vision
- pneumonia
- allergic reaction
If you think you are having an allergic reaction to Bortezomib Juno, tell you doctor immediately or go to Accident and Emergency at your nearest hospital.
Symptoms usually include some or all of the following:
- rash, itching or hives on the skin
- shortness of breath, wheezing or difficulty breathing
- swelling of the face, lips, tongue or other parts of the body
Other side effects not listed above may also occur in some people.
Tell your doctor, nurse or pharmacist if you notice any other effect that is making you feel unwell.
Product description
Storage
Bortezomib Juno should be kept in a cool dry place, protected from light, where the temperature stays below 25°C.
What it looks like
Bortezomib Juno is a white to off-white powder in a glass vial.
Each pack contains one single-use vial.
Before injection, Bortezomib Juno powder is dissolved in a small quantity of sterile, sodium chloride solution.
The solution for injection is clear and colourless.
Bortezomib Juno 3.5 mg vial (AUST R 283343)
Bortezomib Juno 2.5 mg vial (AUST R 303979)
Bortezomib Juno 1 mg vial (AUST R 283340).
Ingredients
Active ingredient:
- bortezomib 3.5 mg (for Bortezomib Juno 3.5 mg Vials)
OR - bortezomib 2.5 mg (for Bortezomib Juno 2.5 mg Vials)
OR - bortezomib 1.0 mg (for Bortezomib Juno 1.0 mg Vials)
Other ingredients:
- mannitol (E 421)
- nitrogen
Sponsor
Juno Pharmaceuticals Pty Ltd
42 Kelso Street,
Cremorne,
VIC 3121
Telephone: 1800 620 076
This leaflet was prepared in: January 2021.
Published by MIMS July 2021



 For additional information concerning melphalan and prednisone, see respective product information.
For additional information concerning melphalan and prednisone, see respective product information.
 In addition, when Bortezomib Juno is given in combination with other chemotherapeutic medicinal products, appropriate dose reductions for these medicinal products should be considered in the event of toxicities, according to the recommendations in the respective Product Information documents.
In addition, when Bortezomib Juno is given in combination with other chemotherapeutic medicinal products, appropriate dose reductions for these medicinal products should be considered in the event of toxicities, according to the recommendations in the respective Product Information documents.

 Solutions prepared this way have been shown to be chemically stable for these periods. However, from a microbiological point of view, the solution should be used immediately. If storage is necessary, hold at 2°C and 8°C for no longer than 24 hours before discarding.
Solutions prepared this way have been shown to be chemically stable for these periods. However, from a microbiological point of view, the solution should be used immediately. If storage is necessary, hold at 2°C and 8°C for no longer than 24 hours before discarding. In the combination study of bortezomib with rituximab, cyclophosphamide, doxorubicin and prednisone (VcR-CAP) in previously untreated mantle cell lymphoma patients, the incidence of thrombocytopenia adverse events (≥ Grade 4) was 32% versus 2% for the rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) arm. The incidence of bleeding adverse events (≥ Grade 3) was 1.7% (4 patients) in the VcR-CAP arm and was 1.2% (3 patients) in the R-CHOP arm.
In the combination study of bortezomib with rituximab, cyclophosphamide, doxorubicin and prednisone (VcR-CAP) in previously untreated mantle cell lymphoma patients, the incidence of thrombocytopenia adverse events (≥ Grade 4) was 32% versus 2% for the rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP) arm. The incidence of bleeding adverse events (≥ Grade 3) was 1.7% (4 patients) in the VcR-CAP arm and was 1.2% (3 patients) in the R-CHOP arm. During consolidation therapy of the GIMEMA study, grade 3 - 4 adverse events were similar to those reported during induction, although rates were much lower. Notably, the rate of grade 3 - 4 peripheral neuropathy was 1.2% with VcTD consolidation compared to 0% with TD consolidation.
During consolidation therapy of the GIMEMA study, grade 3 - 4 adverse events were similar to those reported during induction, although rates were much lower. Notably, the rate of grade 3 - 4 peripheral neuropathy was 1.2% with VcTD consolidation compared to 0% with TD consolidation.

 Although, in general safety data were similar for the IV and SC treatment groups, Table 15 highlights differences larger than 10% in the overall incidence of adverse drug reactions between the two treatment arms.
Although, in general safety data were similar for the IV and SC treatment groups, Table 15 highlights differences larger than 10% in the overall incidence of adverse drug reactions between the two treatment arms. Patients who received bortezomib subcutaneously compared to intravenous administration had 13% lower overall incidence of treatment emergent adverse drug reactions that were grade 3 or higher in toxicity (57% vs 70% respectively; p-value is 0.0784), and a 5% lower incidence of discontinuation of bortezomib (22% vs 27%; p-value is 0.5052). The overall incidence of diarrhoea (24% for the SC arm vs 36% for the IV arm; p-value is 0.0572), gastrointestinal and abdominal pain (6% for the SC arm vs 19% for the IV arm; p-value is 0.0049), asthenic conditions (27% for SC arm vs 39% for IV arm), upper respiratory tract infections (14% SC arm vs 26% IV arm; p-value is 0.0903) and peripheral neuropathy NEC (38% SC arm vs 53% IV arm; p-value is 0.0444) were 12%-15% lower in the subcutaneous group than the intravenous group. In addition, the incidence of peripheral neuropathies that were grade 3 or higher in toxicity was 10% lower (6% for SC vs 16% for IV; p-value is 0.0264), and the discontinuation rate due to peripheral neuropathies was 8% lower for the subcutaneous group (5%) as compared to the intravenous group (14%); p-value is 0.0771.
Patients who received bortezomib subcutaneously compared to intravenous administration had 13% lower overall incidence of treatment emergent adverse drug reactions that were grade 3 or higher in toxicity (57% vs 70% respectively; p-value is 0.0784), and a 5% lower incidence of discontinuation of bortezomib (22% vs 27%; p-value is 0.5052). The overall incidence of diarrhoea (24% for the SC arm vs 36% for the IV arm; p-value is 0.0572), gastrointestinal and abdominal pain (6% for the SC arm vs 19% for the IV arm; p-value is 0.0049), asthenic conditions (27% for SC arm vs 39% for IV arm), upper respiratory tract infections (14% SC arm vs 26% IV arm; p-value is 0.0903) and peripheral neuropathy NEC (38% SC arm vs 53% IV arm; p-value is 0.0444) were 12%-15% lower in the subcutaneous group than the intravenous group. In addition, the incidence of peripheral neuropathies that were grade 3 or higher in toxicity was 10% lower (6% for SC vs 16% for IV; p-value is 0.0264), and the discontinuation rate due to peripheral neuropathies was 8% lower for the subcutaneous group (5%) as compared to the intravenous group (14%); p-value is 0.0771.
 In addition, compared with the TD arm, Progression Free Survival (PFS) was also significantly longer for patients randomized to the Vc-TD arm (HR, 0.629 [CI: 0.451-0.878], p = 0.0061). The estimated 3-year PFS rate was 68% in the VTD arm and 56% in TD (p = 0.0057) (see Figure 1). 58 (24.5%) and 86 (36%) patients progressed or died, respectively. The estimated 3-year probability of progression or relapse was 29% in the Vc-TD versus 39% in the TD arm (HR, 0.609 [CI: 0.425-0.873], p = 0.0073; p = 0.0061 by Kaplan-Meier analysis) (see Figure 2).
In addition, compared with the TD arm, Progression Free Survival (PFS) was also significantly longer for patients randomized to the Vc-TD arm (HR, 0.629 [CI: 0.451-0.878], p = 0.0061). The estimated 3-year PFS rate was 68% in the VTD arm and 56% in TD (p = 0.0057) (see Figure 1). 58 (24.5%) and 86 (36%) patients progressed or died, respectively. The estimated 3-year probability of progression or relapse was 29% in the Vc-TD versus 39% in the TD arm (HR, 0.609 [CI: 0.425-0.873], p = 0.0073; p = 0.0061 by Kaplan-Meier analysis) (see Figure 2).
 The IFM-2005, Phase III, randomised (1:1:1:1), multi-centre, open-label study was conducted to compare the efficacy and safety of bortezomib-dexamethasone (Vc-Dex) and vincristine-doxorubicin-dexamethasone (VAD) as induction therapy prior to HDT-ASCT, and to evaluate the impact of post-induction consolidation therapy. Patients in this study were randomised to receive VAD plus no consolidation (arm A1), VAD plus dexamethasone, cyclophosphamide, etoposide, cis-platin (DCEP) consolidation (arm A2), Vc-Dex plus no consolidation (arm B1), or Vc-Dex plus DCEP consolidation (arm B2).
The IFM-2005, Phase III, randomised (1:1:1:1), multi-centre, open-label study was conducted to compare the efficacy and safety of bortezomib-dexamethasone (Vc-Dex) and vincristine-doxorubicin-dexamethasone (VAD) as induction therapy prior to HDT-ASCT, and to evaluate the impact of post-induction consolidation therapy. Patients in this study were randomised to receive VAD plus no consolidation (arm A1), VAD plus dexamethasone, cyclophosphamide, etoposide, cis-platin (DCEP) consolidation (arm A2), Vc-Dex plus no consolidation (arm B1), or Vc-Dex plus DCEP consolidation (arm B2). A total of 184/218 (84.4%) and 197/223 (88.3%) evaluable patients who received VAD and Vc-Dex induction, respectively, underwent autologous stem cell transplantation. The number of patients who received a second transplantation was 41 (20.8%) in the Vc-Dex arm, compared to 50 (27.2%) for patients in the VAD arm. Post-transplant response rates are shown in Table 19.
A total of 184/218 (84.4%) and 197/223 (88.3%) evaluable patients who received VAD and Vc-Dex induction, respectively, underwent autologous stem cell transplantation. The number of patients who received a second transplantation was 41 (20.8%) in the Vc-Dex arm, compared to 50 (27.2%) for patients in the VAD arm. Post-transplant response rates are shown in Table 19. In addition, the median PFS was 29.7 months among patients who received VAD versus 36.0 months among patients who received Vc-Dex induction, with 128 (52.9%) of 242 and 110 (45.8%) of 240 patients, respectively, having progressed (p = 0.064, or p = 0.057 if adjusted for initial stratification factors) after median follow-up of 31.2 months.
In addition, the median PFS was 29.7 months among patients who received VAD versus 36.0 months among patients who received Vc-Dex induction, with 128 (52.9%) of 242 and 110 (45.8%) of 240 patients, respectively, having progressed (p = 0.064, or p = 0.057 if adjusted for initial stratification factors) after median follow-up of 31.2 months. At the time of a pre-specified interim analysis, the primary endpoint, time to progression, was met and patients in the MP arm were offered VcMP treatment. Survival continued to be followed after the interim analysis. Median follow-up in the initial analysis (Table 21 and Figure 1) was 16.3 months. Median follow-up in the last survival analysis (Figure 2) was 36.7 months. Median overall survival in the MP arm was 43.1 months and was not reached in the VcMP arm. Fifty percent of subjects in the MP arm subsequently received bortezomib.
At the time of a pre-specified interim analysis, the primary endpoint, time to progression, was met and patients in the MP arm were offered VcMP treatment. Survival continued to be followed after the interim analysis. Median follow-up in the initial analysis (Table 21 and Figure 1) was 16.3 months. Median follow-up in the last survival analysis (Figure 2) was 36.7 months. Median overall survival in the MP arm was 43.1 months and was not reached in the VcMP arm. Fifty percent of subjects in the MP arm subsequently received bortezomib. The time to progression (TTP) was significantly longer on the bortezomib arm (see Figure 3).
The time to progression (TTP) was significantly longer on the bortezomib arm (see Figure 3). A significant survival advantage is shown with bortezomib (see Figure 4).
A significant survival advantage is shown with bortezomib (see Figure 4).


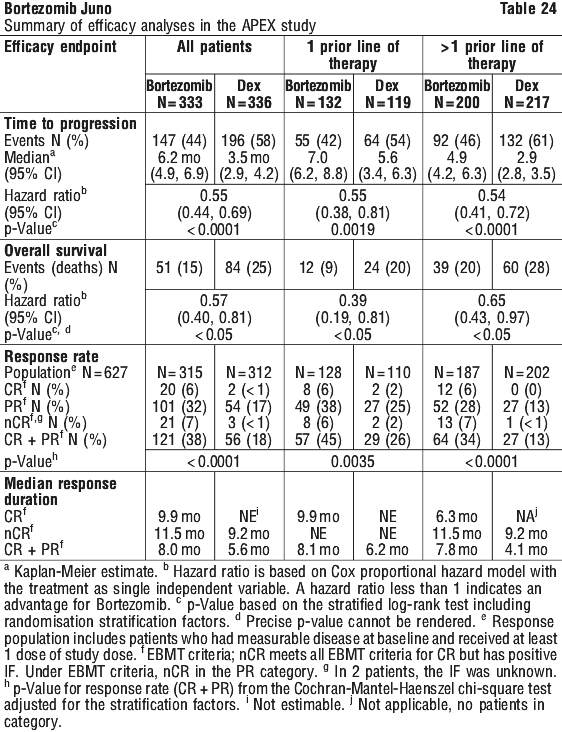 For the 121 patients achieving a response (CR or PR) on the bortezomib arm, the median duration was 8.0 months (95% CI: 6.9, 11.5 months) compared to 5.6 months (95% CI: 4.8, 9.2 months) for the 56 responders on the dexamethasone arm.
For the 121 patients achieving a response (CR or PR) on the bortezomib arm, the median duration was 8.0 months (95% CI: 6.9, 11.5 months) compared to 5.6 months (95% CI: 4.8, 9.2 months) for the 56 responders on the dexamethasone arm. As shown in Figure 6, bortezomib had a significant survival advantage relative to dexamethasone (p < 0.05). The median follow-up was 8.3 months.
As shown in Figure 6, bortezomib had a significant survival advantage relative to dexamethasone (p < 0.05). The median follow-up was 8.3 months.
 Table 26 presents a cross-tabulation summary of best response by algorithm after 4 cycles versus after 8 cycles for patients who received dexamethasone. Eighty-two subjects in the SC treatment group and 39 subjects in the IV treatment group received dexamethasone after cycle 4.
Table 26 presents a cross-tabulation summary of best response by algorithm after 4 cycles versus after 8 cycles for patients who received dexamethasone. Eighty-two subjects in the SC treatment group and 39 subjects in the IV treatment group received dexamethasone after cycle 4. Relative to previously reported outcomes, the ORR after 8 cycles of treatment (52% in both treatment groups) and time to progression (median 10.4 months and 9.4 months in SC and IV treatment groups, respectively), including the effect of the addition of dexamethasone from cycle 5 onwards, were higher than observed in prior registration study with single agent IV bortezomib, APEX, (38% ORR and median TTP of 6.2 months for the bortezomib arm). Time to Progression and ORR was also higher compared to the subgroup of patients on APEX that received only 1 prior line of therapy (43% ORR and median TTP of 7.0 months) (Table 21).
Relative to previously reported outcomes, the ORR after 8 cycles of treatment (52% in both treatment groups) and time to progression (median 10.4 months and 9.4 months in SC and IV treatment groups, respectively), including the effect of the addition of dexamethasone from cycle 5 onwards, were higher than observed in prior registration study with single agent IV bortezomib, APEX, (38% ORR and median TTP of 6.2 months for the bortezomib arm). Time to Progression and ORR was also higher compared to the subgroup of patients on APEX that received only 1 prior line of therapy (43% ORR and median TTP of 7.0 months) (Table 21). Patients who did not obtain an optimal response to therapy with bortezomib alone were able to receive high-dose dexamethasone in conjunction with bortezomib (i.e., 40 mg dexamethasone with each dose of bortezomib administered orally as 20 mg on the day of and 20 mg the day after bortezomib administration, (i.e., Days 1, 2, 4, 5, 8, 9, 11, and 12), thus 160 mg over 3 weeks. Eighteen percent (13/74) of patients achieved or had an improved response (CR 11% or PR 7%) with combination treatment.
Patients who did not obtain an optimal response to therapy with bortezomib alone were able to receive high-dose dexamethasone in conjunction with bortezomib (i.e., 40 mg dexamethasone with each dose of bortezomib administered orally as 20 mg on the day of and 20 mg the day after bortezomib administration, (i.e., Days 1, 2, 4, 5, 8, 9, 11, and 12), thus 160 mg over 3 weeks. Eighteen percent (13/74) of patients achieved or had an improved response (CR 11% or PR 7%) with combination treatment.

